
离子对色谱法测定盐酸达克罗宁中的杂质
2017-04-07 01:29刘明明张根元王浦海
生物加工过程 2017年2期
刘明明,张根元,王浦海
(1.南京工业大学药学院,江苏南京211800; 2.江苏省药物研究所有限公司,江苏南京211800)
离子对色谱法测定盐酸达克罗宁中的杂质
刘明明1,张根元2,王浦海2
(1.南京工业大学药学院,江苏南京211800; 2.江苏省药物研究所有限公司,江苏南京211800)
采用Kromasil C18色谱柱,以磷酸盐缓冲液(称取辛烷磺酸钠2.34 g及KH2PO46.8 g,加H2O 1 L使溶解,用H3PO4调节pH至3.0):甲醇(体积比40∶ 60)为流动相,流速1.0 mL/min,检测波长282 nm,柱温25 ℃。杂质A、B与盐酸达克罗宁在0.05~0.5 μg/mL范围内线性关系良好。杂质A、B的平均回收率分别为99.66%和105.1%,相对标准偏差(RSD)分别为2.12%和4.17%。本方法为盐酸达克罗宁杂质的检查提供了科学依据。
盐酸达克罗宁;离子对色谱法;杂质
盐酸达克罗宁(Dyclonine hydrochloride,1),化学名为1-(4-丁氧基苯)-3-(1-哌啶基)-1-丙酮盐酸盐,是一种起效快且安全性高的局部麻醉药[1-2]。中国药典尚未收载本品,美国药典第35版规定以L13柱测定其含量,国内也有采用C18柱进行测定的报道,但这些方法均是用有机相-磷酸二氢钾[4-7]或有机相-三乙胺[8-9]体系作为流动相,且鲜见其相关杂质检测的报道。根据本品的结构特征及合成工艺路线[10],推测最终产品中可能存在的工艺杂质包括:4-羟基苯乙酮(起始原料,杂质A)和4-丁氧基苯乙酮(中间体,杂质B);降解杂质包括:1-(4-丁氧基苯基)-2-丙烯-1-丙酮(杂质C)和1-(4-羟基苯基)-2-丙烯-1-丙酮(杂质D),见图1。
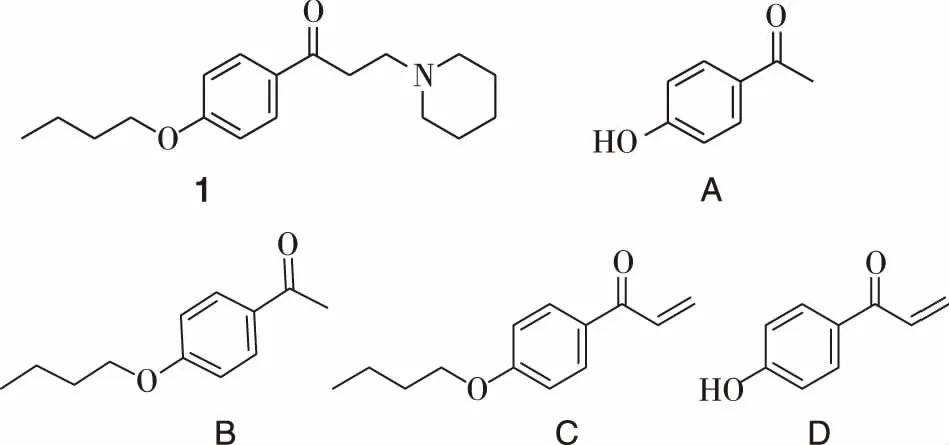
1—盐酸达克罗宁;A—4-羟基苯乙酮;B—4-丁氧基苯乙酮;C—1-(4-丁氧基苯基)-2-丙烯-1-丙酮;D—1-(4-羟基苯基)-2-丙烯-1-丙酮图1 盐酸达克罗宁(1)及相关杂质Fig.1 Structures of dyclonine hydrochlorido (1) and its related substances
在有机相的体系下色谱检测盐酸达克罗宁的主峰峰形拖尾严重,且相关杂质分离不佳。为了弥补现有方法的不足,本文建立了一种新的离子对色谱法,在流动相中引入离子对试剂辛烷磺酸钠,使主峰峰形及主成分与杂质的分离度均得到极大改善。该方法专属性强、灵敏度高,可适用于盐酸达克罗宁相关杂质的检查。考虑到本品中实际存在A、B两个已知杂质,杂质C、D仅在强制降解条件下出现,故本研究在新的离子对色谱条件下仅对杂质A、B进行方法学研究。
1 材料与方法
1.1 材料与仪器
盐酸达克罗宁(1)对照品(中国生物制品药品检定研究院,批号20150402, 含量99.94%),盐酸达克罗宁(1)样品(江苏省药物研究所有限公司,批号20150803,20150901,20150903);有关物质A(江苏省药物研究所有限公司,批号:BH-AB02993-150302,含量:99.71% );有关物质B(江苏省药物研究所有限公司,批号:20150701,含量:99.56%);辛烷磺酸钠(分析纯),德国默克公司;KH2PO4(分析纯),南京化学试剂有限公司;甲醇(色谱纯),霍尼韦尔贸易(上海)有限公司;H3PO4(分析纯),南京化学试剂有限公司;水为自制超纯水。
LC-2010A型高效液相色谱仪,日本Shimadzu公司;Micromass ZQ型液质联用仪,美国Waters公司。
1.2 实验方法
1.2.1 检测波长的选择
取1及有关物质A、B适量,分别用流动相溶解并稀释制成每1 mL中约含5 μg的溶液,照紫外-可见分光光度法(中国药典2015年版通则0401)测定,在200~400 nm波长范围内扫描,结果1及有杂质A、B 均在282 nm左右的波长处有最大吸收,故选择282 nm作为本品杂质的检测波长。
1.2.2 色谱条件
色谱柱为Kromasil C18硅烷键合硅胶柱(250 mm×4.6 mm,5 μm);流动相:磷酸盐缓冲液(称取辛烷磺酸钠2.34 g及KH2PO46.8 g,加水1 L使溶解,用H3PO4调节pH至3.0,摇匀)-甲醇(体积比为40∶ 60);检测波长282 nm;流速1.0 mL/min;柱温25 ℃;进样量20 μL。
1.2.3 系统适用性溶液的配制
精密称取1 10 mg,置50 mL量瓶中,加适量流动相溶解,置水浴中加热4 h,取出后放冷,加流动相稀释至刻度,摇匀即得。
1.2.4 杂质混合溶液的配制
精密称取A、B各10 mg,置于100 mL量瓶中,流动相定容,精密量取5 mL,置50 mL量瓶中,用流动相稀释至刻度,摇匀即得。
1.2.5 杂质对照溶液的配制
精密量取有关物质混合溶液1 mL,置于100 mL量瓶中,用流动相稀释至刻度,摇匀即得。
1.2.6 供试品溶液的配制
精密称取1 20 mg,置于100 mL量瓶中,加流动相适量溶解,并加流动相稀释至刻度,摇匀即得。
1.2.7 对照溶液的配制
精密量取供试品溶液1 mL,置于100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取1 mL,置于10 mL量瓶中,用流动相稀释至刻度,摇匀即得。
2 结果与讨论
2.1 分离度
取系统适用性溶液20 μL,注入液相色谱仪,记录色谱图,如图2所示。由图2可知:1与相关杂质的分离度均大于2.0,理论板数以1计算,大于4 000。
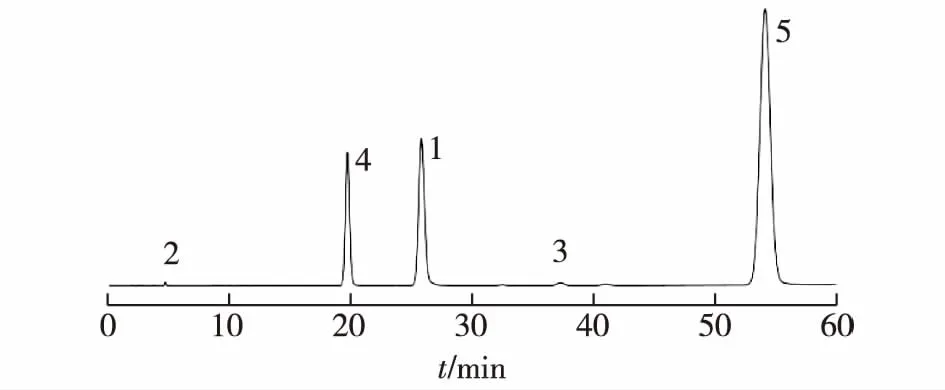
1—1;2—杂质A;3—杂质B;4—杂质C;5—杂质D图2 系统适用性试验溶液色谱图Fig.2 Chromatogram of the system suitability test solution
2.2 专属性试验
取样品1 10 mg,共6份,各置50 mL量瓶中,振摇使其溶解,分别进行强酸、强碱、氧化、光照和高温破坏试验,另一份不作破坏,作对照试验,其中强酸、强碱样品进样前须中和,结果见图3。由图3可知:在上述色谱条件下,降解产物与1分离良好,且经苛刻条件下破坏后的主峰峰纯度符合要求(纯度因子大于峰纯度阈值)。
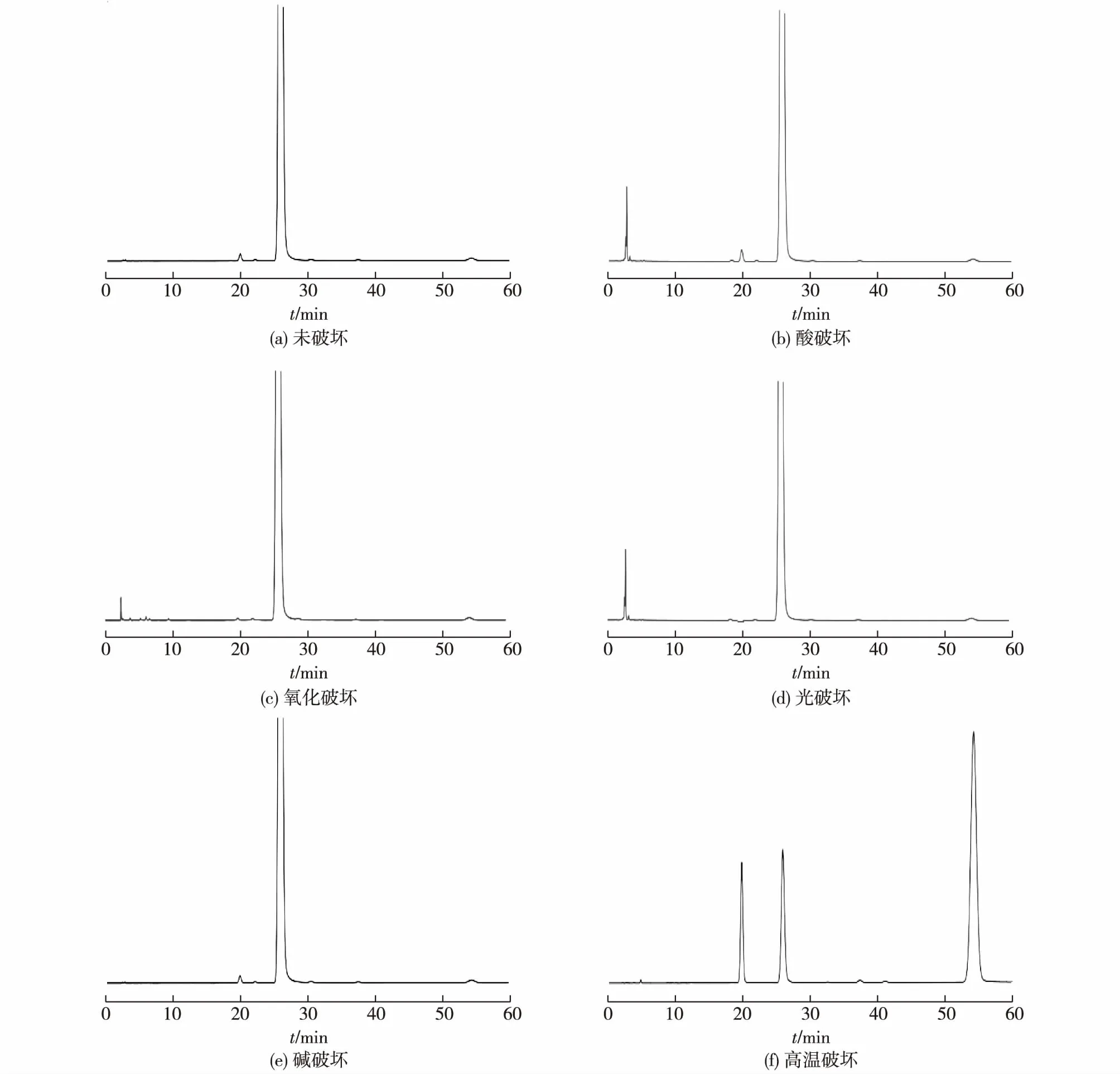
图3 专属性试验色谱图Fig.3 HPLC chromatograms of forced specificity test
由图3可知:本品1在高温条件下不稳定,降解出了2个主要杂质C和D,该2个主要杂质在已有文献报道的检测方法中与主成分难以分离,而在本文色谱条件下与主成分分离良好,说明本色谱条件对本品与相关杂质的分离效果有极大改善。
2.3 线性试验及校正因子测定
精密称取1及杂质A、B对照品适量,用流动相溶解并定量稀释制成浓度为0.05~0.5 μg/mL的1及A、B的混合对照品溶液,作为线性测定系列浓度溶液,分别进样20 μL,记录色谱图。并以浓度ρ为横坐标,峰面积A为纵坐标,绘制标准曲线。同时根据1及杂质A、B线性方程的斜率,计算A、B的校正因子,结果见表1。

表1 线性试验结果
2.4 重复性试验
取样品1,平行配制供试品溶液及对照溶液6份,取20 μL分别进样,记录色谱图。按加校正因子自身对照法计算有关物质,结果6份样品有关物质A含量的RSD为0.69%、有关物质B含量的RSD为1.48%、单个杂质的相对标准偏差(RSD)为1.22%、总杂质的RSD为1.08%。
2.5 回收率试验
分别精密称取1 10 mg共9份,各置50 mL量瓶中,分别精密加入有关物质混合溶液0.5、1.0和1.5 mL,即得50%、100%和150%杂质限度的混合液,各3份,分别进样测定,计算回收率。结果有关物质A、B的平均回收率为99.66%和105.1%,RSD分别为2.12%和4.17%(n=9)。
2.6 精密度与稳定性试验
取 1及有关物质对照溶液,分别连续进样6次,记录峰面积,计算得1及有关物质A、B峰面积的RSD分别为0.85%、1.35%和1.40%。
取1供试品溶液,分别配制后于0、2、4、6、8、12和24 h进样,记录色谱图及峰面积,结果有关物质A、B及1峰面积的RSD分别为0.97%、1.46%和0.80%,未见其他杂质出现,说明供试品溶液放置24 h稳定。
2.7 样品测定
取3批样品1供试品、对照溶液,分别进样,记录色谱图。按加校正因子的自身对照法计算相关杂质,结果见表2。
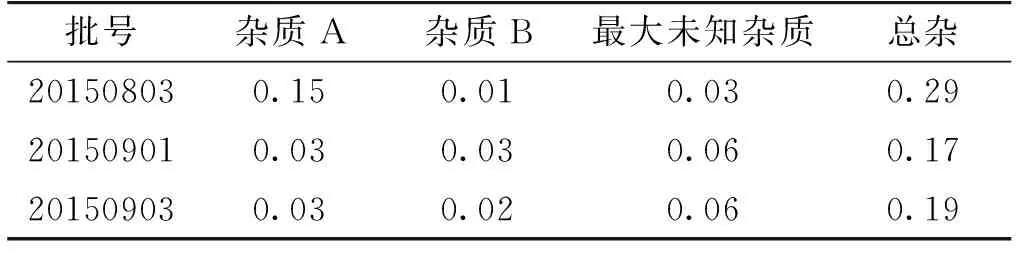
表2 相关杂质测定结果
由表2可知:盐酸达克罗宁样品的相关杂质都被有效检测出来。因为本品1为一含碱性氮原子的极性化合物,在反相色谱条件下与固定相相互作用较强,导致其拖尾严重,峰型差柱效低,且与相邻杂质分离不佳。反离子对试剂辛烷磺酸钠的引入,与本品结构中的碱性氮原子相结合形成中性分子的离子对化合物,使得其疏水性增强,与固定相的作用减弱,很好地改善了峰型,极大地提高了柱效,与相关杂质的分离度明显增加,从而解决了已有文献报道的方法中盐酸达克罗宁峰拖尾严重、分离度差的问题。
3 结论
本文在参考国内外已报道的盐酸达克罗宁检测方法文献的基础上,对色谱条件进行了改善,建立了测定本品杂质更为合理、科学的离子对色谱检测法。通过调整离子对试剂种类、用量、溶液pH及有机相比例,经优化筛选,最终确立了本方法,为盐酸达克罗宁杂质的检查提供了科学依据。
[1] 张丽君,朱晶,祝修权.盐酸达克罗宁的合成.广东化工,2010,37(8):106-108.
[2] 张小瑞,王金波,慕高萌.1%盐酸达克罗宁凝胶的制备及质量控制.实用药物与临床,2015,18(1):66-69.
[3] The United States Pharmacopoeia Commission Inc.The United States Pharmacopoeia.(USP35).Rockville:the United States Pharmacopoeia Convention,Inc.,2012:2997.
[4] 于桂兰,杨建春,张琦,等.HPLC法同时测定敏宁搽剂中二组分含量.药物分析杂志,2011,31(5):977-979.
[5] 毛宗惠.HPLC法测定复方盐酸达克罗宁乳膏中盐酸达克罗宁含量.世界最新医学信息文摘,2015,15(18):36-37.
[6] 张勇,黄爱文,宋洪涛.高效液相色谱法测定多效胃镜乳中主要成分含量.中国药业,2014,23(20):57-59.
[7] 谭璐,张广求,刘文.HPLC法同时测定复方乳酸左氧氟沙星烧伤凝胶中两组分的含量.中国药房,2013,24(40):3833-3835.
[8] 傅峰,施孝金,郁韫超.复方地塞米松涂膜中醋酸地塞米松和盐酸达克罗宁的HPLC法测定.中国医药工业杂志,2012,43(11):935-937.
[9] 李成,颜晗,罗意文,等.HPLC测定复方盐酸达克罗宁薄荷脑润肤止痒水中达克罗宁的含量.海南医学,2010,21(14):125-126.
[10] 孙田江,陆宏国,周斌,等.一种盐酸达克罗宁合成方法:101544616 B.2011-05-25.
(责任编辑 周晓薇)
Determination of impurities in Dyclonine hydrochloride by ion pair chromatography
LIU Mingming1,ZHANG Genyuan2,WANG Puhai2
(1.College of Pharmaceutical Engineering,Nanjing Tech University,Nanjing 211800,China;2.Jiangsu Provincial Institute of Material Medica Co.Ltd., Nanjing 211800,China)
A C18column was used,with the mobile phase of phosphate buffer (dissolved octane sulfonic acid sodium salt 2.34 g and KH2PO46.8 g in 1 L water, pH adjusted to 3.0 by H3PO4):methanol (V/V=40∶ 60) with flow rate 1.0 mL/min at 25 ℃ with the detection wavelength of 282 nm.It was linear for the related substances A,B and Dyclonine hydrochloride in the concentration ranges of 0.05-0.5 μg/mL.The average recoveries of A and B were 99.66% and 105.12%,with RSDs of 2.12% and 4.17%,respectively. This mothod provides a scientific basis for the determination of the Dyclonine hydrochloride substances.
Dyclonine hydrochloride;ion pair chromatography;related substances
10.3969/j.issn.1672-3678.2017.02.008
2016-05-12
刘明明(1991—),女,江苏南京人,研究方向:药物制剂及分析;张根元(联系人),副研究员,E-mail:zhanggenyuan63@126.com
R927.1
A
1672-3678(2017)02-0045-04
猜你喜欢
人人健康(2021年14期)2021-08-06
西南国防医药(2021年3期)2021-05-14
天津医科大学学报(2021年1期)2021-01-26
中国医药科学(2020年1期)2020-03-02
时代邮刊(2019年22期)2019-12-17
时代邮刊·下半月(2019年11期)2019-09-22
山东化工(2018年15期)2018-09-20
中国现代医生(2017年28期)2017-11-17
首都食品与医药(2015年18期)2015-11-03
药学研究(2012年2期)2012-10-25
