
DNA barcodes for identification of closely related species in Camellia
2017-03-31 01:10WENBei-bei,DENGLi,YEXia等
广东农业科学 2017年1期
DNA barcodes for identification of closely related species in Camellia
The Camellia genus containing about 280 species is the only genus with great commercial values in the family Theaceae. The products of this genus such as seed-oil,ornamental plants and different kinds of tea are popular in the current market. The classification of species in Camellia genus using molecular methods may have potential power in finding cryptic species,rearing new variety and controlling the trade involving counterfeit. DNA barcoding is regarded as a promising tool for species-level identification marked by short,standardized DNA sequences. In this study,we tested the value of widely used barcoding markers(trnH-psbA and matK)on 64 Camellia species and compared with those of other genera in different family downloaded from NCBI. In all,61 trnH-psbA sequences and 61 matK sequences were obtained and the pairwise distances,aligned length,variable sites were calculated. As the consistency of matK sequences was extremely high(98.40%),we mainly focused on further analysis of trnH-psbA region. In genus level,all 61 species were successfully distinguished from 7 other genera. When comes to species level within Camellia genus,the discrimination rate was only 13.11%. The results indicate that trnH-psbA region may have effective ability in genus-level identification,but has weak practicability in specieslevel discrimination.
Camellia;trnH-psbA;matK;DNA barcodes;species identification
Camellia including 280 species[1]has great economical importance in the family Theaceae[2]and has the general characteristics of alternate leaves,large flowers,numerous stamens,a short radical and broad cotyledons[3]. There exist several famous members of this genus because of their special economic values[1]. For example,some species can produce high quality oils from their seeds[4],and a few species with magnificent showy flowers like C. japonica,C. sasanqua and C. reticulata are widely cultivated in the world[2],and the most famous species is C. sinensis(L.)O. Ktze.,known as the common tea plant,which can product various types of tea containing rich catechins[5].
At present,the general agreed taxonomic revisions are Sealy's[6],Chang Hung-da's[7]and Min T L's[8]taxonomy system. Sealy J R reported that genus Camellia was classified into 82 species of 12 sects and about 20 species were unclear as the lack of information and references. After general investigation,Chang Hung-da rearranged the system in 4 subgenera,20 sects,280 species. MIN T L accepted Chang's classification and added some new species in this genus. Recently,several molecular strategies have been used for species identification and genetic polymorphism analysis such as RAPD(Random Amplification Polymorphic DNA),AFLP(AmplifiedFragments Length Polymorphism)and ISSR(Inter-simple Sequence Repeat)[9].
DNA barcoding has been reported as a new molecular tool for species identification using short,standardized DNA regions[10-11]and is already used for discovering new species and assign unknown species to their correct position[12]. There is already a universal barcode for most animal lineages,which is mitochondrial cytochrome c oxidase 1(CO1)gene sequence[10,13]. However,the power of CO1 for plant identification is weak due to the low rate of mutation[14-15]. Useful DNA barcoding loci requires conversed flanking region for easy amplification and sufficient internal variability for species discrimination[16]. Although,several candidates of plant DNA barcodes have been proposed and tested in different families,genera,including coding and non-coding regions[12,16],still no single marker can fulfill all the traits of DNA barcoding in plant Kingdom[17-18].
The plastid intergenic spacer trnH-psbA was used as potential promising DNA region to identify the flowering plants by Kress[12]. Researchers also recommended the combination of trnH-psbA and coding rbcL gene as a two-locus global barcodes for their universality and the ability of species discrimination[18]. Many results have suggested trnH-psbA as a useful DNA barcode for its high rate of amplification and sequencing in previous researches[19-20]. In this study,the power of trnH-psbA and matK regions was tested as the makers for molecular identification and 64 species from the genus Camellia were sampled,to evaluate the capacity of trnH-psbA and matK in discrimination and provide a molecular solution of taxonomy in Camellia genus.
1 Materials and methods
1.1 Plant samples
We collected 64 specimens including 16 sections of Camellia from the International Camellia Species Garden in Jinhua,Zhejiang,China(Table 1). Branches were taken from the plants and cultivated in water. After getting back to the laboratory,we froze the leaves immediately and stored the samples at -80℃.
1.2 DNA extraction,amplification and sequencing
Total DNA was extracted for each sample from approximately 50 mg of freeze-dried leaves using a modified cetyltrimethylammonium bromide(CTAB)procedure. DNA extraction of each sample was hydrated in 50 μL 1.0×TE(pH=8.0),then transferred to 1.5 mL centrifuge
tube and stored at -80℃. The forward and reverse primers used in this study were listed in Table 2. After diluting to 50 ng/μL with water of each DNA extraction,the PCR amplifications for trnH-psbA and matK were performed in 50 μL reaction mixtures containing 25 μL 2×Taq Master Mix(Tiangen Biotech,Beijing,CN),1 μL template DNA(30-50 ng/μL)and distilled water to volume. The PCR cycling conditions were as follows:94℃5 min,(94℃ 30 s,58℃ 30 s,72℃ 1 min)for 35 cycles,72℃ 7 min,4℃ for ever[12]. The PCR products were electrophoresed with SYBR Green I on 1.5% agarose gel in 1.0 TAE(Tris-acetate)buffer(Fig.1),and purified using the Sangon Purification Kit(Sangon,Shanghai,China),and then sequenced in both directions with the PCR primers on an ABI 3730 automated sequencer.

(续Table 1)
1.3 Data analysis
Bidirectional sequences were aligned with Clustal W[21]and the alignments were adjustedmanually with the use of CHROMAS LIFE 2.0 software. GC content,aligned length,variable sites and parsimony-information(PI)sites,pairwise genetic distances of two regions were analyzed using MEGA 5.0[22]Kimura two-parameter(K2P)model. The consistency of all sequences was calculated in DNAman 6(Lynnon Biosoft). Blast and tree-based methods were used to evaluate the identification power of trnH-psbA and matK primer pairs.
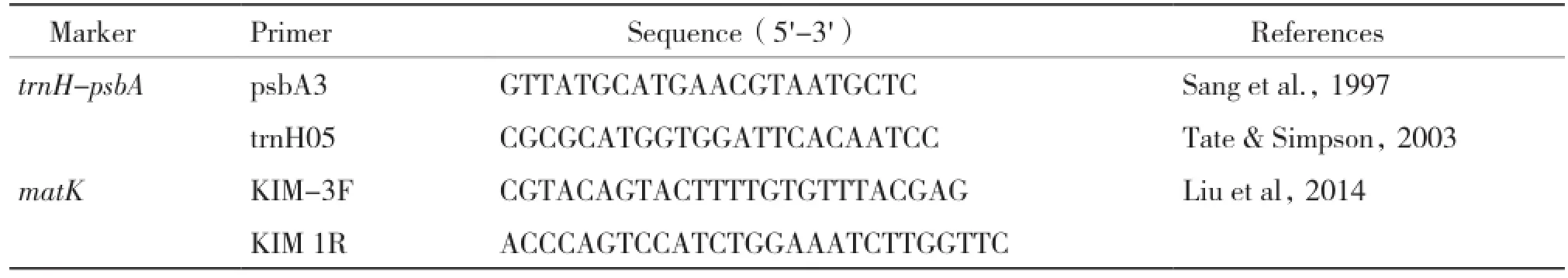
Table 2 The primer sequences used for each sample of Camellia
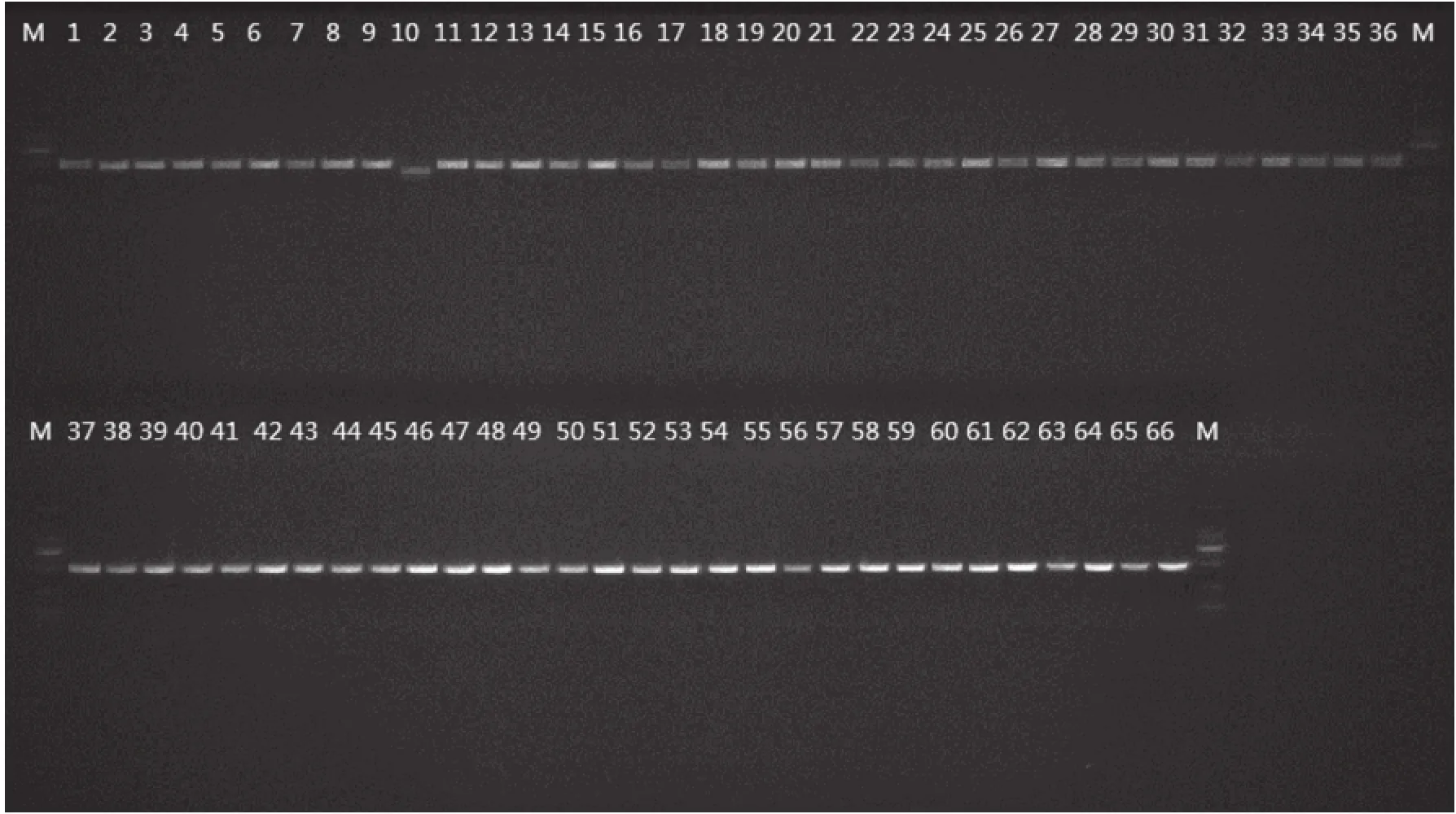
Fig.1 Electrophoregram of PCR products of Camellia
The trnH-psbA sequences of 3 genera in Theaceae and 6 other genera which were listed in Table 3 with accession number,species name,genera,length and references,and mapped in Fig. 2 were blasted and downloaded from the National Center for Biotechnology Information(NCBI)nucleotide database(http://www.Ncbi.nlm.nih.gov/ BLAST/).
Neighbor-joining(NJ)trees were constructed using MEGA 5 in Kimura two-parameter(K2P)model based on 1 000 bootstrap replicates. Species with a bootstrap value above 60% were considered as successful identifications[23-24].
2 Results and analysis
2.1 PCR amplification and sequencing
TrnH-psbA showed 100% success rate in PCR amplification and 95.31% in sequencing. MatK performed 100% in amplification and 95.31% in sequencing. In all,61 trnH-psbA and 61 matK sequences for 16 sects genera of Camellia species were submitted to GenBank(trnH-psbA:KX121705-KX121765;matK:KX216412-KX216472.)
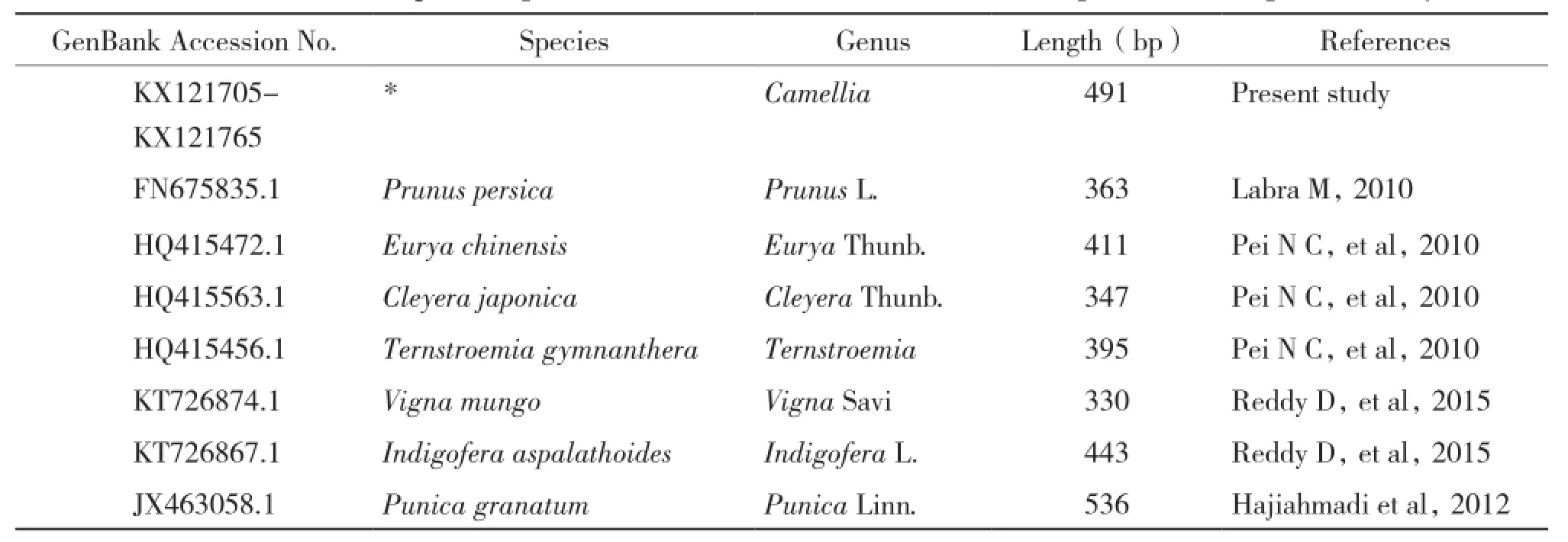
Table 3 List of trnH-psbA sequences downloaded from GenBank and sequenced in the present study
2.2 Alignment and identification
The aligned length of trnH-psbA(excluding external primers)was 491 bp with 67 variable sites(13.64%),400 conserved sites(81.47%)and 31 parsimony-information sites(6.31%)and they were listed in Table 4 with the information of matK sequences. The mean nucleotide level for all obtained trnH-psbA sequences was as follows:A=31.63%,C=10.98%,G=17.83%,T=39.57%.
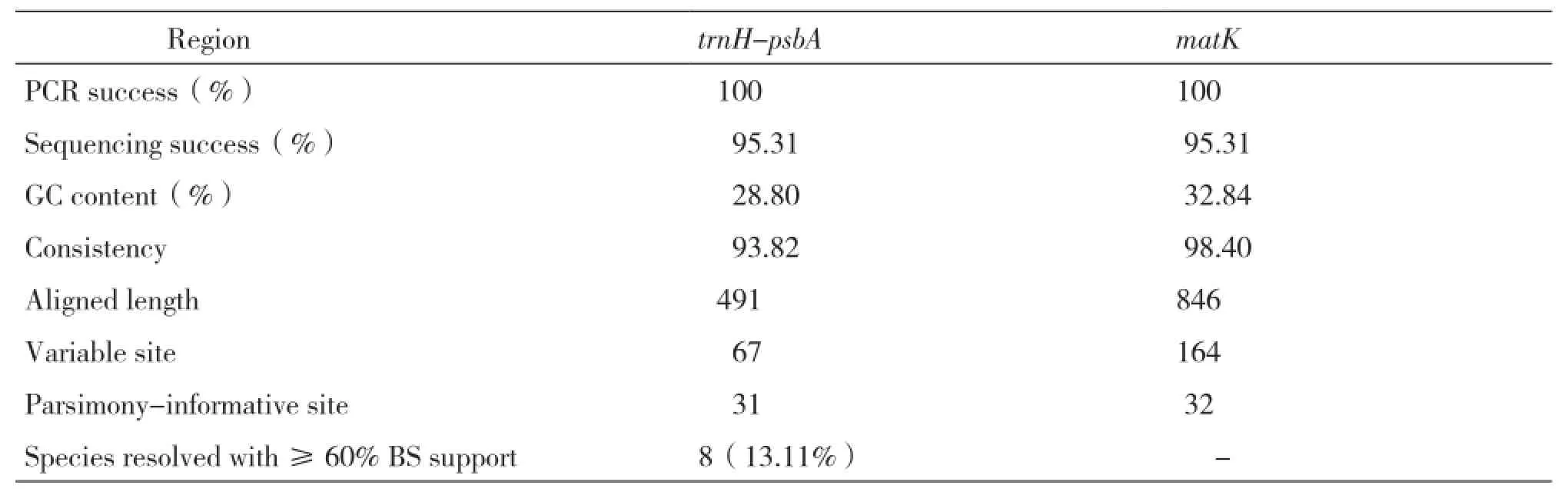
Table 4 Summary statistics for trnH-psbA and matK sequences of Camellia
Using blast searching in GenBank with the 61 trnH-psbA and matK sequences of 16 Sect genera,it showed that all the sequences can be classified to their high homologous sequences. The consistency of trnH-psbA and matK sequences was 93.82% and 98.40% performed by DNAman.
The trnH-psbA region showed higher interspecific divergence compared with other researchers results’[19-20]. The highest pairwise distance of trnH-psbA sequences in 61 species was 0.034 for two groups,and those were C. kissi Wall. and C. albogigas Hu,C. albogigas Hu and C. lanceoleosa Chang et Chiu ex Chang et Ren. The lowest pairwise distance was 0.000 for six groups,and those were C. jingyunshanica Chang et J. H. Xiong and C. nitidissima Chi,C. jingyunshanica Chang et J. H. Xiong and C. kwangtungensis Chang,C. nitidissima Chi and C. kwangtungensis Chang,C. fengchengensis Liang et Zhong and C. rhytidocarpa Chang et Liang,C. longipetiolata(Hu)Chang et Fang and C. euphlebia Merr. ex Sealy. As to pairwise distances of 16 sects genera,the highestwas 0.025,which was between Sect Archecamellia Sealy and Sect stereocarpus Pierre Sealy. The lowest was 0.009 for five groups of sects genera which were as follows:Sect Oleifera Chang and Sect Longipedicellata Chang,Sect Oleifera Chang and Sect Furfuracea Chang,Sect Longipedicellata Chang and Sect Chrysantha Chang,Sect Longipedicellata Chang and Sect Furfuracea Chang,Sect Camellia L Dyer and Sect Glaberrima Chang(Table 5).

Table 5 A pairwise comparison of the trnH-psbA sequences about 16 sects genera Camellia,as calculated by MEGA 5.0[22]
For genera identification,trnH-psbA exhibited high level of discrimination rate(100%),which can clearly identify samples from different genera. For species identification,there were 8 of the 61 species(13.11%)successfully discriminated in NJ tree(Fig. 2).
It contains 61 samples of Camellia genus and 6 other genera downloaded from NCBI. Successfully distinguished species(Bootstrap values≥60%)are marked triangles.
3 Discussion and conclusion
3.1 PCR and sequencing success
As important criteria for DNA barcoding[18],PCR and sequencing success rates are essential factors to evaluate the practicability of candidate barcodes. In present study,the trnH-psbA region showed high level of primer universality,with 100% in PCR amplification and 95.31% in sequencing. This performance is consistent with previousresearches that have reported success rates >95%[19-20]. Many results suggested that the matK region was difficult to amplify and sequence and it usually required several pairs of primers[16-25]. However,in present research,matK region reached 100% in PCR success rate and 95.31% in sequencing with only a pair of primer which was higher than the published result(85.4% in amplification of matK[20]). This indicated the universality of the chosen primers for both regions was pretty high and suitable for Camellia genus.

Fig 2 Neighbor-joining tree based on the trnH-psbA sequences
3.2 Identification of genera and species
The crucial ability for a barcode is to recover monophyletic clusters of samples from a single species to provide correct identification[15]. Although the discrimination of closely related species is still a challenge for DNA barcoding[26]. CBOL Plant Working Group has suggested thatmatK region is as one of the core plant barcodes for its rapid evolutionary and high rate of species discrimination published in recent studies(CBOL Plant Working Group,2009). However,it is found that the consistency of matK region in Camellia genus is extremely high(98.40%)and it is hardly to identify samples among species.
TrnH-psbA performed rather poorly in species discrimination,only 8 of 61 samples(13.11%)have been successfully identified as reported in Solidago(NJ,BS:17%)and Curcuma(NJ,BS:15.94%)[16,20]. The 8 species that had been identified successfully by NJ tree(BS≥60%)were as follows:C. liberistyloides Chang,C. yunnanensis(Pitard)Coh. st,C. taliensis(W. W. Sm.)Melch,C. assamica(mast.)Chang,C. subintegra Huang et Shi,C. japonica Linn,C. longganensis C. F. Liang et S. L. Mo,C. longgangensis var. Patens S. L. mo et Y. C. Zhong. Previous results showed that the species recovery ability of trnH-psbA was pretty unstable,which can be 3.7% to 80.40%. This indicated that using trnH-psbA to identify individual species of Camellia is not an effective solution.
In present study,the identification rate among genera of trnH-psbA region reaches 100%. All 61 species of Camellia genus are successfully distinguished(Bootstrap values,BS:82%)from the genera downloaded from NCBI. TrnH-psbA region is easy to amplify and sequence,and has high level of genera discrimination rate showed in present work. Therefore,it implies that trnH-psbA can be a practical candidate DNA barcode for plants in different genera.
3.3 Taxonomic implications in Camellia genus
The most heated debated purposes of DNA barcoding are species discovery and identification[27]. Although species discrimination in Camellia was low based on DNA barcoding,this method exhibited a strong identification power at genera,section and subgenus levels[28]. For most species in Camellia,the interrelationships remained unresolved. Therefore,it was reasonable that the correct identified samples were quite few. Recently,the complete chloroplast genome of Camellia sinensis cv. Longjing 43 have been sequenced[29],more highly variable plastid DNA markers can be tested and suggested for identifying Camellia species,and it’s possible that suitable barcodes will be developed in the future.
When it comes to practical situation of identification and new species discovery,molecular methods may require the combination with traditional taxonomy to accomplish the purposes. Identifying unknown organisms using molecular markers needs an existing database and can only be established by the delimitation of traditional taxonomy. To identify new species,only taking the barcoding sequences as evidences is not enough,an expert in taxonomy is more helpful.
Given the fact that DNA barcoding provides a rapid diagnostic means for sorting samples genetically,it can help with the process of delimiting species boundaries and expanding the taxonomic databases[30]. In present study,the DNA sequence database and the relationships among species based on DNA barcoding provided information for further taxonomic study of Camellia species.
Acknowledgements
The authors thank:SHAO Fu-sheng from the Institution of International Camellia Species Garden(Jinhua,Zhejiang,China)for helping us in obtaining permits and in-city assistance;MENG Li,WANG Hui-yu,ZHAO Zhi-hao(College of Forestry,Henan Agricultural University)for providing the molecular biology laboratory;LI Hui(College of Horticulture,Nanjing Agriculture University)for his help with the data analysis.
[1] Chang H T,Bartholomew B. Camellias[M].Portland,Oregon:Timber Press,1984:304.
[2] Bezbaruah H P. Cytological investigations in the family Theaceae-I. Chromosome numbers in some Camellia species and allied genera[J]. Caryologia,1971,24(4):421-426.
[3] Takhtajan A. Diversity and Classification of Flowering Plants[M]. New York:Columbia University Press,1998:191-192.
[4] Liu Y,Mo R,Zhong D,et al. The fate of organophosphorus pesticides during Camellia oil production[J]. Journal of Food Science,2015,80(8):1926-1932.
[5] Ajay R,Harsh P S,Ashu G. Investigation of major phenolic antioxidants from Camellia sinensis fruits[J]. Cogent Chemistry,2015,1:1080652.
[6] Sealy J R. A Revision of the Genus Camellia[M]. London:The Royal Hort. Soc.,1958.
[7] Chang H T. Thea-A section of beveragial teatrees of the genus Camellia[J]. Acta Scientiarum Naturalium Universitatis Sunyatseni,1981,(1):87-99.
[8] Wu Z Y,Raven P H. Flora of China[M]. Beijing:Science Press,2007.
[9] Pan L Q. The application of several molecular technologies in the genus Camellia. Chinese[J]. Horticulture Abstracts,2015,4:53-56.
[10] Hebert P D N,Cywinska A,Ball S L,et al. Biological identifications through DNA barcodes[J]. Proceedings of the Royal Society B:Biological Sciences,2003,270:313-321.
[11] Hebert P D N,Penton E H,Burns J M,et al. Ten species in one:DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator[J]. USA:Proceedings of the National Academy of Sciences,2004,101:14 812-14 817.
[12] Kress W J,Wurdack K J,Zimmer E A,et al. Use of DNA barcodes to identify flowering plants[J]. USA:Proceedings of National Academy of Sciences,2005,102:8369-8374.
[13] Janzen D H,Hajibabaei M,Burns J M,et al. Wedding biodiversity inventory of a large and complex Lepidoptera fauna with DNA barcoding[J]. Philosophical Transactions of the Royal Society B:Biological Sciences,2005,360:1835-1845.
[14] Hollingsworth P M,Graham S W,Little D P. Choosing and using a plant DNA barcode[J]. PLoS One,2011,6:e19254.
[15] Fazekas A J,Burgess K S,Kesanakurti P R,et al. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well[J]. PLoS One,2008,3:e2802.
[16] Hollingsworth M L,Andra C A,Forrest L L,et al. Selecting barcoding loci for plants:evaluation of seven candidate loci with species level sampling in three divergent groups of land plants[J]. Molecular Ecology Resources,2009,9:439-457.
[17] Newmaster S G,Fazekas A J,Steeves R A D,et al. Testing candidate plant barcode regions in the Myristicaceae[J]. Molecular Ecology Resources,2008,8:480-490.
[18] Kress W J,Erickson D L. A two-locus global DNA barcode for land plants:The coding rbcL gene complements the non-coding trnH-psbA spacer region[J]. PLoS One,2007,2(6):e508.
[19] Chen J,Zhao J T,David L,et al. Testing DNA barcodes in closely related species of Curcuma(Zingiberaceae)from Myanmar and China[J]. Molecular Ecology Resources,2015,15:337-348.
[20] Thompson J D,Higgins D G,Gibson T J. CLUSTAL W:improving the sensitivity of progressive multiple sequence alignment through sequence weighting,position-specific gap penalties and weight matrix choice[J]. Nucleic Acids Research,1994,22:4673-4680.
[21] Tamura K,Peterson D,Peterson N,et al. MEGA 5:mmolecular evolutionary geneticsanalysis using maximum likelihood,evolutionary distance,and maximum parsimony methods[J]. Molecular Biology and Evolution,2011,28:2731-2739.
[22] Cai Z M,Zhang Y X,Zhang L N,et al. Testing four candidate barcoding markers in temperate woody bamboos(Poaceae:Bambusoideae)[J]. Journal of Systematics and Evolution,2012,50(6):527-539.
[23] Krawczyk K,Szczeci nska M,Sawicki J. Evaluation of 11 singlelocus and seven multilocus DNA barcodes in Lamium L.(Lamiaceae)[J]. Molecular Ecology Resources,2013,14:272-285.
[24] Ford C S,Ayres K L,Toomey N,et al. Selection of candidate coding DNA barcoding regions for use on land plants[J]. Botanical Journal of the Linnean Society,2009,159:1-11.
[25] Ragupathy S,Newmaster S G,Murugesan M,et al. DNA barcoding discriminates a new cryptic grass species revealed in an ethnobotany study by the hill tribes of the Western Ghats in southern India[J]. Molecular Ecology Resources,2009,9:164-171.
[26] Desalle R. Species discovery versus species identification in DNA barcoding efforts:response to Rubinoff[J]. Conservation Biology,2006,20:1545-1547.
[27] Yan L J,Liu J,Michael M,et al. DNA barcoding of Rhododendron(Ericaceae),the largest Chinese plant genus in biodiversity hotspots of the Himalaya-Hengduan Mountains[J]. Molecular Ecology Resources,2014,4:932-944.
[28] Ye X Q,Zhao Z H,Zhu Q W,et al. Entire chloroplast genome sequence of tea(Camellia sinensis cv. Longjing 43):a molecular phylogenetic analysis[J]. Journal of Zhejiang University(Agriculture and Life Sciences),2014,40(4):404-412.
[29] Jing G U,Su J X,Lin R Z,et al. Testing four proposed barcoding markers for the identification of species within Ligustrum L.(Oleaceae)[J]. Journal of Systematics and Evolution,2011,49(3):213-224.
[30] Zhang C Y,Wang F Y,Yan H F,et al. Testing DNA barcoding in closely related groups of Lysimachia L.(Myrsinaceae)[J]. Molecular Ecology Resources,2012,12:98-108.
(责任编辑 储霞玲)
WEN Bei-bei,DENG Li,YE Xia,HE Wei,LIU Jian-jun,SU Hui,LI Mei-feng,FENG Jian-can
(College of Horticulture,Henan Agricultural University,Zhengzhou 450002,China)
DNA条形码在山茶属近缘植物鉴别中的应用
温贝贝,邓 丽,叶 霞,贺 巍,刘建军,苏 会,李美凤,冯建灿
(河南农业大学园艺学院,河南 郑州 450002)
山茶属(Camellia)是山茶科(Theaceae)中物种丰富、经济效益较高的属。分类学上对山茶属的亚属、组以及种的划分争议较大,著名的3大山茶属植物分类系统分别由Sealy、张宏达和闵天禄提出。DNA条形码技术是指通过对植物基因组中的特定基因进行片段扩增、测序发现其碱基变化规律的技术手段,在分类学研究中显示出巨大的应用潜力。选取trnH-psbA和matK序列的通用引物,对山茶属不同植物基因组DNA进行扩增和序列测定,分别得到了ttrnH-psbA序列和matK序列各61条,山茶属matK序列相似度极高(98.40%),物种分辨率较低。trnH-psbA的属间物种分辨能力达到100%,而在山茶属内物种分辨率仅为13.11%。结果表明:trnH-psbA序列能够有效地区分不同属间的植物,但种间分类能力较弱。
山茶属;trnH-psbA;matK;DNA条形码;物种分辨率
S682.31 Document Code:A Article ID:1004-874X(2017)01-0055-11
Received date:2016-10-08
Project funded:Science and technology innovation team support plan of Henan university(14IRTSTHN011)
Author:WEN Bei-bei(1990-),female,postgraduate,E-mail:2672802468@qq.com
WEN Bei-bei,DENG Li,YE Xia,et al. DNA barcodes for identification of closely related species in Camellia[J]. Guangdong Agricultural Sciences,2017,44(1):55-65.
猜你喜欢
课堂内外·教师版(2022年4期)2022-05-23
少年文艺·开心阅读作文(2021年8期)2021-09-05
广东园林(2020年3期)2020-07-03
文苑(2020年6期)2020-06-22
小学科学(学生版)(2019年5期)2019-05-21
大观(书画家)(2018年6期)2018-07-08
学苑创造·B版(2017年10期)2017-12-21
中央民族大学学报(自然科学版)(2016年2期)2016-06-27
中央民族大学学报(自然科学版)(2015年3期)2015-06-11
学生天地·初中(2014年1期)2014-02-17
