
甘蔗线条花叶病毒HC-Pro基因的分子变异分析
2017-03-29 03:11李文凤张志想李世访
植物保护 2017年2期
贺 振, 李文凤, 张志想, 李世访*
(1. 扬州大学园艺与植物保护学院, 扬州 225009; 2. 中国农业科学院植物保护研究所/植物病虫害生物学国家重点实验室, 北京 100193; 3. 云南省农业科学院甘蔗研究所/云南省甘蔗遗传改良重点实验室, 开远 661699)
研究报告
甘蔗线条花叶病毒HC-Pro基因的分子变异分析
贺 振1,2, 李文凤3, 张志想2, 李世访2*
(1. 扬州大学园艺与植物保护学院, 扬州 225009; 2. 中国农业科学院植物保护研究所/植物病虫害生物学国家重点实验室, 北京 100193; 3. 云南省农业科学院甘蔗研究所/云南省甘蔗遗传改良重点实验室, 开远 661699)
为了解析甘蔗线条花叶病毒Sugarcanestreakmosaicvirus(SCSMV)不同分离物HC-Pro基因的分子变异规律,本研究利用RT-PCR法扩增获得SCSMV HC-Pro基因的序列,通过生物信息学分析,分别从重组、系统发生、选择压力等方面研究SCSMV HC-Pro基因的分子变异特征。共测定了44条SCSMV HC-Pro基因序列,相似性最低值为70%;HC-Pro基因重组频率较低,仅发现3个重组位点,其中一个系首次报道;与先前报道相比,部分新测定云南蔗区的SCSMV分离物在HC-Pro基因上形成一个新组-第Ⅲ组;HC-Pro基因处于很强的负选择压力作用,未发现正向选择作用位点。本研究结果进一步证明SCSMV HC-Pro基因具有高度的遗传多样性。
甘蔗线条花叶病毒; HC-Pro基因; 分子变异
甘蔗线条花叶病毒Sugarcanestreakmosaicvirus(SCSMV)是近年来从甘蔗花叶病株上鉴定出的一种新病原,属于马铃薯Y病毒科Potyviridae禾本科病毒属Poacevirus[1-2]。该病毒分子量大小约为10 kb,编码一个多聚蛋白,经水解酶切割后可产生10个成熟的蛋白质,在P3蛋白氨基端有一个PIPO蛋白[2-3]。自然条件下,该病毒可侵染甘蔗和高粱,目前尚未发现SCSMV的传毒介体[1-3]。2012年,我们在云南省甘蔗产区首次发现该病毒,经调查发现,SCSMV在云南省甘蔗主产区发生越来越普遍,部分地区呈现流行暴发趋势[1, 4-5]。因此设计合理的SCSMV防治策略对于抑制其在中国蔗区传播流行具有十分重要的意义。RNA病毒的分子进化研究有利于我们加深对于病毒传播途径、流行路线以及寄主相互适应方式等[6-7]的研究,从而为设计合理的病毒病害防治策略提供依据。目前,针对SCSMV的研究较少。Viswanathan等[8]和Bagyalakshmi等[9]发现SCSMV印度分离物存在较高的遗传多样性。我们的前期研究发现SCSMV具有一个典型的“准种”结构,普遍存在同种病毒不同分离物或株系混合侵染的现象[5];SCSMV依据不同基因可划分为9个组(第Ⅰ~第Ⅸ组),并且存在重组现象[5]。不同于Potyvirus,HC-Pro基因是SCSMV基因组中遗传多样性最高的一个基因[5],前期研究中由于基因序列的限制,无法对其进行一个系统的分子进化分析,因此,本研究试图选取不同地区的多个典型分离物,在重组、系统发生、选择压力等方面,对其进行系统全面的分子进化分析。
1 材料与方法
1.1 病毒分离物
2009-2012年间从云南省7个甘蔗主产区和国家甘蔗种质资源圃内采集到具有典型花叶症状(图1)的甘蔗样品324份。新鲜叶片样品经RT-PCR法检测鉴定,将SCSMV阳性样品冷冻干燥后,-80℃保存备用。本研究中所用样品详细信息见表1。
1.2 克隆测序
根据GenBank已公布的SCSMV序列保守区,设计用于扩增HC-Pro基因的引物SCSM-HC-Pro-F (5′-TGGACTCATTTGACGCCAGG-3′)和SCSM-HC-Pro-R(5′-CGCTAACCTTGTTGTGTCGT-3′),预期扩增的目的片段大小约为1 500 bp。引物由上海生工生物工程有限公司合成。
采用TRIzol试剂法提取SCSMV甘蔗叶片中的总RNA,提取方法参照试剂盒说明书进行。取2 μL总RNA,采用Promega公司MLV反转录试剂盒,用反向引物SCSM-HC-Pro-R进行反转录获得cDNA。以cDNA为模板进行PCR。PCR扩增采用50 μL反应体系:10×PCR buffer 5 μL,dNTPs (2.5 mmol/L) 4 μL,SCSM-HC-Pro-F (10 μmol/L) 2 μL,SCSM-HC-Pro-R (10 μmol/L) 2 μL,ddH2O 34.5 μL,longTaqpolymerase (5 U/μL) 0.5 μL,cDNA 2 μL。PCR反应条件为:94℃预变性5 min;94℃变性30 s,55℃复性30 s,72℃延伸1 min,共30个循环;最后一轮循环后72℃延伸10 min。4℃保存。PCR反应结束后,吸取产物2 μL进行1%的琼脂糖凝胶电泳检测。PCR产物纯化后克隆至pGEM-T载体上,并转化至大肠杆菌Escherichiacoli感受态细胞DH5α中。经菌落PCR鉴定获得阳性重组质粒,将筛选获得的阳性克隆随机选择3~6个送至北京六合华大基因科技股份有限公司测序。测序结果首先通过峰图及序列比对分析排除由PCR扩增引起的突变,然后通过Bioedit 5.0.9拼接完成。
1.3 重组分析
本研究中,经测序所得SCSMV HC-Pro基因的核苷酸序列共44条(GenBank登录号分别为:KU314329~KU314372),BLAST检索结果显示其与麦类花叶病毒Triticummosaicvirus(TriMV)具有最近的亲缘关系。因此,本研究选择TriMV分离物(NC012779)对应的HC-Pro基因序列作为序列比对分析的外组(outgroup)[10]。结合GenBank中所有可用的SCSMV HC-Pro基因的核酸序列形成一个具有106条序列的数据集合。对该数据集合进行比对,过程如下:首先将核酸序列数据对应的氨基酸序列应用CLUSTRAL X2[11]和TRANSALIGN(由Georg Weiller教授惠赠)比对,以保证经过序列比对后得到的核酸序列能够正确地编译出氨基酸序列。经过序列比对后,得到的删除gap后的HC-Pro基因的序列长度为990个核苷酸(nucleotide,nt)。对比对所得序列数据,利用Datamonkey(http:∥www.datamonkey.org/)中GARD和RDP 4.0软件包[12-13]中的RDP[12]、GENECONV[14]、BOOTSCAN[15]、MAXCHI[16]、CHIMAERA[17]、3SEQ[18]和SISCAN[19]7个程序进行重组检测,以发现可能存在的重组位点。在RDP 4.0检测时,各软件参数采用默认值,Bonferroni校正的P为0.05或者0.01,当某分离物有至少3个软件的检测结果P<1.0×10-6时,支持该分离物为重组体[20-21]。在这些分析中,重组体序列会与非重组体序列存在部分相似,为了方便说明,我们将此非重组体作为该重组体的“亲本分离物”(parental isolates),据此,当重组体与亲本分离物位于同一组时,将此重组体命名为组内重组体(intralineage recombinant);当重组体与亲本分离物位于非同一组时,则命名为组间重组体(interlineage recombinant)[20, 22]。最后,将SCSMV序列数据中外组TriMV序列删除,去除TriMV对SCSMV序列造成的gap影响,直接检测确认SCSMV的HC-Pro基因区的重组位点。
表1 本研究中的SCSMV样品采集信息
Table 1 Information ofSugarcanestreakmosaicvirussamples in this study
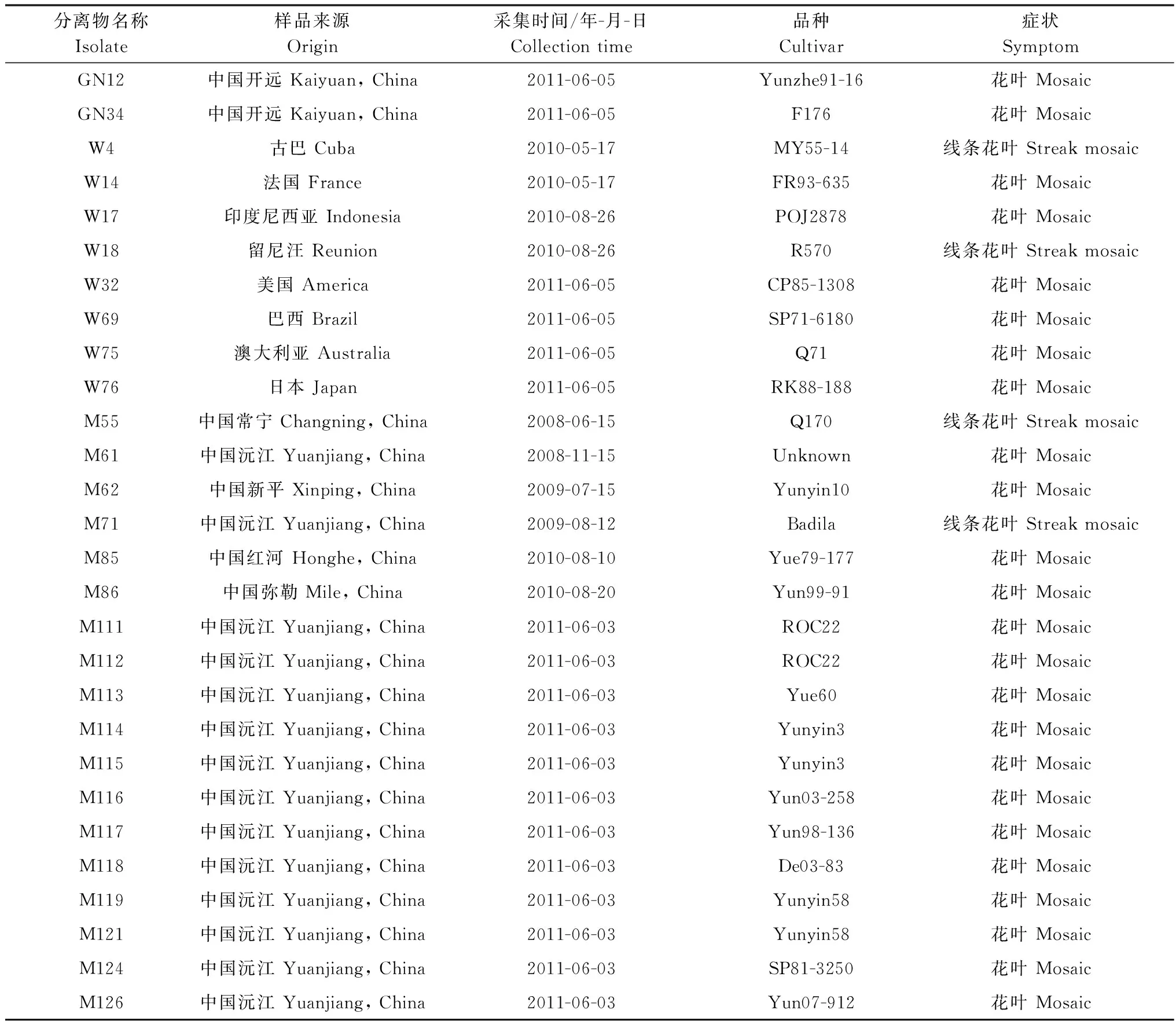
分离物名称Isolate样品来源Origin采集时间/年-月-日Collectiontime品种Cultivar症状SymptomGN12中国开远Kaiyuan,China2011-06-05Yunzhe91-16花叶MosaicGN34中国开远Kaiyuan,China2011-06-05F176花叶MosaicW4古巴Cuba2010-05-17MY55-14线条花叶StreakmosaicW14法国France2010-05-17FR93-635花叶MosaicW17印度尼西亚Indonesia2010-08-26POJ2878花叶MosaicW18留尼汪Reunion2010-08-26R570线条花叶StreakmosaicW32美国America2011-06-05CP85-1308花叶MosaicW69巴西Brazil2011-06-05SP71-6180花叶MosaicW75澳大利亚Australia2011-06-05Q71花叶MosaicW76日本Japan2011-06-05RK88-188花叶MosaicM55中国常宁Changning,China2008-06-15Q170线条花叶StreakmosaicM61中国沅江Yuanjiang,China2008-11-15Unknown花叶MosaicM62中国新平Xinping,China2009-07-15Yunyin10花叶MosaicM71中国沅江Yuanjiang,China2009-08-12Badila线条花叶StreakmosaicM85中国红河Honghe,China2010-08-10Yue79-177花叶MosaicM86中国弥勒Mile,China2010-08-20Yun99-91花叶MosaicM111中国沅江Yuanjiang,China2011-06-03ROC22花叶MosaicM112中国沅江Yuanjiang,China2011-06-03ROC22花叶MosaicM113中国沅江Yuanjiang,China2011-06-03Yue60花叶MosaicM114中国沅江Yuanjiang,China2011-06-03Yunyin3花叶MosaicM115中国沅江Yuanjiang,China2011-06-03Yunyin3花叶MosaicM116中国沅江Yuanjiang,China2011-06-03Yun03-258花叶MosaicM117中国沅江Yuanjiang,China2011-06-03Yun98-136花叶MosaicM118中国沅江Yuanjiang,China2011-06-03De03-83花叶MosaicM119中国沅江Yuanjiang,China2011-06-03Yunyin58花叶MosaicM121中国沅江Yuanjiang,China2011-06-03Yunyin58花叶MosaicM124中国沅江Yuanjiang,China2011-06-03SP81-3250花叶MosaicM126中国沅江Yuanjiang,China2011-06-03Yun07-912花叶Mosaic
1.4 系统发生分析
对上述处理后的序列分别利用PhyML 3.0[23]中的最大似然法(maximum-likelihood,ML)、MEGA 6.0[24]中的邻接法(neighbour-joining,NJ)以及SPLITSTREE 4.11.3[25]中的邻接网法(neighbor-net,NN)进行系统发生分析。在ML法分析中,通过jModeltest 0.1.1[26]分析确定HC-Pro数据的最适核苷酸替代模型为GTR+I+Г4。在ML和NJ法分析中,支长皆用自举法(bootstrap)进行1 000次模拟复制计算检验。系统发育树由TREEVIEW[27]展示。核酸和氨基酸相似性分析分别依据Kimura two-parameter method[28]和Dayhoff PAM 001matrix[29]方法计算,种群内多样性分析由MEGA 6.0[24]计算。在本研究中,通过计算HC-Pro基因的dN/dS值(非同义突变和同义突变之间的比值)来预测该基因所承受的选择压力。计算基于ML法,通过以下两种方法进行检测。首先,利用Datamonkey(http:∥www.datamonkey.org/)中SLAC(single-likelihood ancestor counting)、FEL(fixed-effects likelihood)和REL(random-effects likelihood)在线检测不同位置密码子的选择压力;第二,在MEGA 6.0[24]中利用Pamilo-Bianchi-Li method[30]计算系统树不同分支上密码子的选择压力。当dN/dS<1时,该组分离物处于纯化或负向选择压力下;当dN/dS=1时,说明该组分离物处于中性选择压力中;当dN/dS>1时,说明该组分离物受正向选择或多样化选择作用。
1.5 多样性分析
利用DnaSP 5.0[31]估测不同种群分离物的核酸多样性(nucleotide diversity)和单体型多样性(haplotype diversity)。核酸多样性是指分离物序列间的平均差异;单体型多样性是指样本中单体型出现的频率和数量。一般而言,植物RNA病毒种群的单体型多样性的值较高,核酸多样性的值较低[5, 22, 32-35]。SCSMV的HC-Pro基因的核酸和氨基酸间的多样性分布图可通过SDT 1.0[36]中的Clustal W[11]法计算获得。
2 结果分析
2.1 SCSMV HC-Pro基因的序列分析
2009-2012年对云南7个甘蔗主产地州和国家甘蔗种质资源圃内甘蔗花叶病中SCSMV的发生情况进行了调查分析,结果发现:SCSMV在云南省甘蔗种植区平均发病率30%(96个阳性样品);在甘蔗产区,SCSMV在沅江、开远、新平、常宁、红河和弥勒等县区分布较为广泛[4];在资源圃内,部分品种的甘蔗种质中SCSMV具有较高的检出率(59.1%)。
在SCSMV阳性样品中,选取28个具有典型花叶、线条花叶症状(图1)的样品,对其HC-Pro基因克隆测序。测序结果显示,每个样品中所得不同克隆的序列相似性很高,选取其中差异性较大的序列(44条)登录到GenBank数据库中。去掉引物序列,本研究所获得的HC-Pro基因区段序列长度为1 002 nt。在SCSMV的HC-Pro基因中,并没有发现在马铃薯Y病毒属病毒的HC-Pro基因中广泛存在的KITC、GE、FRNK和PTK等结构域。

图1 甘蔗花叶病症状Fig.1 Symptoms of the sugarcane mosaic disease
2.2 重组分析结果
将本研究所测定的以及在GenBank中获取的共计105个SCSMV HC-Pro基因序列进行重组分析。在利用NN法构建的网状树中,部分分离物之间存在明显的序列交叉,表明部分SCSMV分离物在HC-Pro基因中存在明显的重组现象(数据未显示)。利用RDP 4.0[13]分析发现,在HC-Pro基因中共有3个明显的重组位点,分别位于SCSMV基因组(位点依据SCSMV-ID分离物,登录号: JF488066)的第1 575、1 736和2 273位点,其中,前两个位点在先前的研究中有过报道,2 273位点是本研究中新发现的一个重组位点(表 2)。在105个SCSMV序列中,共发现4个明显的重组体,分别为M117-CL2、M117-CL3、M117-CL7和CB419。本研究所获得的SCSMV序列中没有发现明显的重组位点。
表2 在SCSMV的HC-Pro基因上发生的重组事件1)
Table 2 Recombination event in the HC-Pro gene of SCSMV
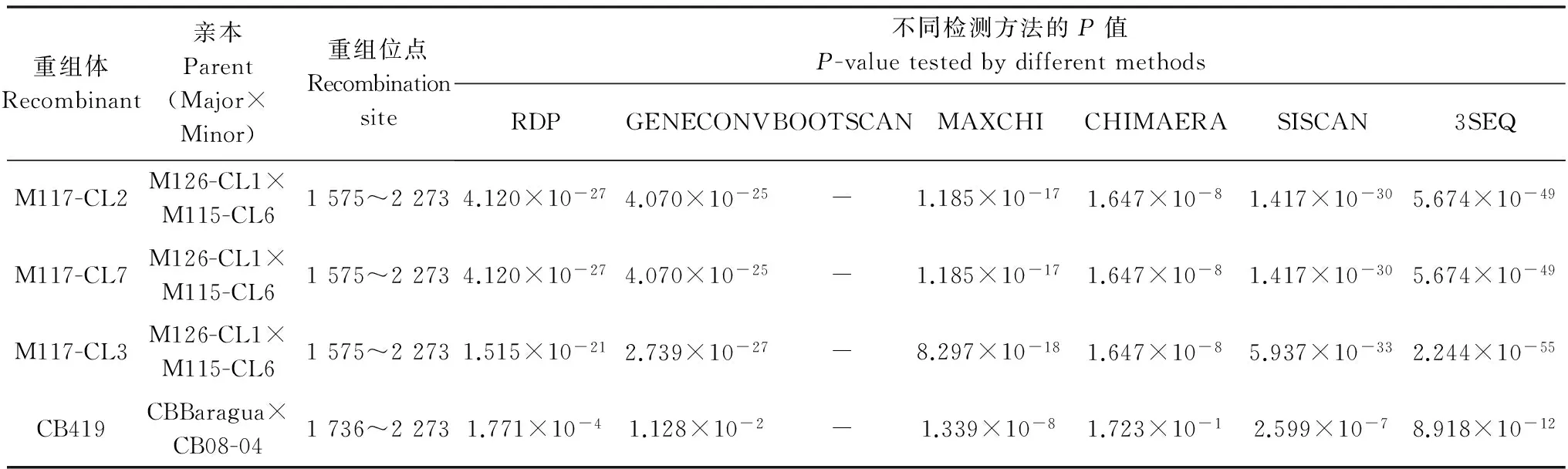
重组体Recombinant亲本Parent(Major×Minor)重组位点Recombinationsite不同检测方法的P值P-valuetestedbydifferentmethodsRDPGENECONVBOOTSCANMAXCHICHIMAERASISCAN3SEQM117-CL2M126-CL1×M115-CL61575~22734.120×10-274.070×10-25-1.185×10-171.647×10-81.417×10-305.674×10-49M117-CL7M126-CL1×M115-CL61575~22734.120×10-274.070×10-25-1.185×10-171.647×10-81.417×10-305.674×10-49M117-CL3M126-CL1×M115-CL61575~22731.515×10-212.739×10-27-8.297×10-181.647×10-85.937×10-332.244×10-55CB419CBBaragua×CB08-041736~22731.771×10-41.128×10-2-1.339×10-81.723×10-12.599×10-78.918×10-12
1) 核苷酸位点依据SCSMV-ID分离物(GenBank登录号: JF488066)。 Regions and sites in accordance to the SCSMV ID isolates (GenBank accession no. JF488066).
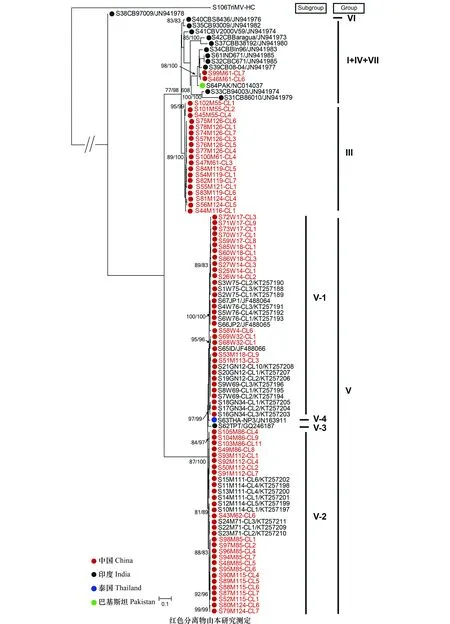
图2 利用ML法对SCSMV HC-Pro基因的系统发生分析Fig.2 Phylogenetic analysis of the helper-component proteinase (HC-Pro) gene sequences ofSugarcane streak mosaic virus using ML method
2.3 系统发育分析
将上述数据集合去掉重组体,对剩余的SCSMV序列进行系统发生分析,ML法和NJ法构建的系统发育树具有相似的拓扑结构,ML树如图2所示。在先前的报道[5]中,SCSMV依据不同基因至少可分为9个组(第I~第IX组),而依据HC-Pro基因包含第Ⅴ、Ⅵ、Ⅶ和第Ⅰ+Ⅳ组。在本研究中,所有分离物共分为4个组,分别为第Ⅲ、Ⅴ、Ⅵ和第Ⅰ+Ⅳ+Ⅶ组,其中第Ⅴ组又可以分为4个亚组(Ⅴ-1、Ⅴ-2、Ⅴ-3和Ⅴ-4)。SCSMV不同地区具有较为明显的地理特异性,其中中国分离物主要集中在第Ⅲ和第Ⅴ组,而印度分离物主要集中在第Ⅵ和第Ⅰ+Ⅳ+Ⅶ组。本研究所得的SCSMV分离物在系统树中广泛分布,且部分来自于云南甘蔗产区的样品形成一个新组-第Ⅲ组。SCSMV在HC-Pro基因的dN/dS为0.21,没有发现正向选择作用位点;不同种群间的dN/dS值远小于1,其中第Ⅲ组具有最小的dN/dS值(0.036),说明SCSMV不同种群都处于较强的负选择压力作用下。
2.4 多样性分析结果
利用SDT 1.0[36]统计计算SCSMV在HC-Pro基因上的核苷酸多样性分布,结果如图3所示。所有SCSMV分离物序列在HC-Pro基因上的核苷酸多样性程度很高,相似性值低至70%(图3)。在系统发生所分的4个组中,第Ⅲ组具有较高的单体型多样性(0.982±0.022)和最低的核苷酸多样性(0.011 21±0.001 25)(表3)。相对于SCSMV印度分离物,在中国SCSMV不同分离物间遗传相似性程度更高(表3)。
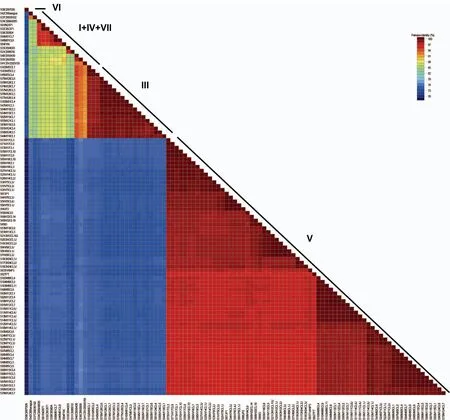
图3 SCSMV HC-Pro基因序列一致率分布图Fig.3 Distribution of pairwise identity scores of the helper-component proteinase (HC-Pro) gene ofSugarcane streak mosaic virus
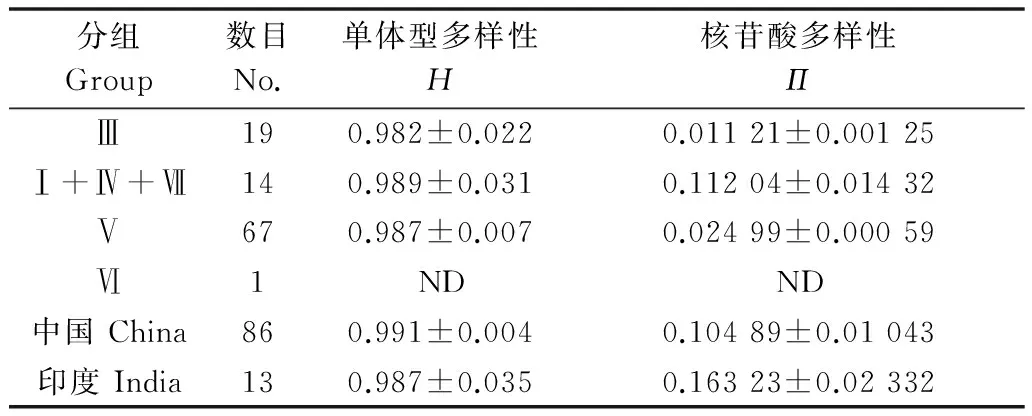
分组Group数目No.单体型多样性H核苷酸多样性ΠⅢ190.982±0.0220.01121±0.00125Ⅰ+Ⅳ+Ⅶ140.989±0.0310.11204±0.01432Ⅴ670.987±0.0070.02499±0.00059Ⅵ1NDND中国China860.991±0.0040.10489±0.01043印度India130.987±0.0350.16323±0.02332
1)Π: 依据序列样本中碱基间的平均差异计算。ND: 未测定。Π: Nucleotide diversity was estimated by the average pairwise difference between sequences in a sample, based on all sites. ND: Not detected.
3 结论与讨论
目前,已经报道了多种植物病毒分子进化与种群结构的研究,特别是Potyvirus,包括PVY[33, 37-38]、TuMV[20, 22]以及TVBMV[35]等。我们先前对SCSMV的分子进化研究发现,HC-Pro基因是其基因组中遗传多样性最高的基因,但限于分离物数量有限,未能作充分的研究[5]。在本研究中,我们从云南省甘蔗产区以及国家甘蔗种质资源圃内选择了28个具有典型甘蔗花叶症状的样品,测定了其HC-Pro基因序列,结合GenBank中已报道的序列信息,对其进行系统的分子变异与种群特征分析。植物病毒在每个单独的寄主中,是以一个“突变云”或者“准种”[39]方式存在的。因此,经单斑分离获得单一纯化的生物克隆是进行分子变异和种群结构分析的必要条件。而在部分植物病毒中,由于没有单斑寄主,无法排除不同基因组片段间错误拼接的可能,因此通常集中于分析病毒的某些单一扩增片段[4-5]。在本研究中,我们通过单一扩增SCSMV的HC-Pro基因,测定不同克隆序列的方式进行分析,以确保不存在可能的拼接错误。
重组是遗传多样性的重要来源,是促进病毒进化的主要动力之一。在PVY[32]和TuMV[20, 22]等Potyvirus中,HC-Pro基因的重组发生频率很高。而在本研究中,SCSMV的HC-Pro基因中仅发现3个重组位点,这表明SCSMV HC-Pro基因中发生重组的频率很低。He等[5]的研究中,由于分离物的数量限制,SCSMV依据HC-Pro基因可分为4个组,分别为第Ⅴ、Ⅵ、Ⅶ和第Ⅰ+Ⅳ组。本研究中测定的部分源自云南蔗区的分离物序列形成一个新组,对照SCSMV P1基因的系统树,将其定名为第Ⅲ组。第Ⅲ组SCSMV分离物的发现,增加了HC-Pro基因与P1和CP基因在系统发生分析上的一致性。SCSMV不同组间具有清晰的地理特异性,中国分离物主要集中在第Ⅲ组和第Ⅴ组。本研究测定的SCSMV资源圃和甘蔗产区分离物位于不同分支,遗传差异较大,具有很明显的亚组结构,这表明云南甘蔗产区和资源圃内SCSMV可能具有不同的起源。通常,大部分的动植物病毒处于强的负选择压力下。本研究中,HC-Pro基因的dN/dS远小于1,表明SCSMV在此基因上受到强的负选择压力作用,这可能是与其基因功能的稳定性相适应的。核苷酸多样性分布结果与系统发生结果相一致,进一步证明SCSMV HC-Pro基因具有高度的遗传多样性,而该结果可能与其功能相适应,尽管目前尚不明确SCSMV HC-Pro的基因功能。
[1] Xu D L, Zhou G H, Xie Y J, et al. Complete nucleotide sequence and taxonomy ofSugarcanestreakmosaicvirus, member of a novel genus in the familyPotyviridae[J]. Virus Genes, 2010, 40(3): 432-439.
[2] Hema M, Sreenivasulu P, Savithri H S. Taxonomic position ofSugarcanestreakmosaicvirusin the familyPotyviridae[J]. Archives of Virology, 2002, 147(10): 1997-2007.
[3] Li Wenfeng, He Zhen, Li Shifang, et al. Molecular characterization of a new strain ofSugarcanestreakmosaicvirus(SCSMV)[J]. Archives of Virology, 2011, 156(11): 2101-2104.
[4] He Zhen, Li Wenfeng, Yasaka R, et al. Molecular variability ofSugarcanestreakmosaicvirusin China based on an analysis of the P1 and CP protein coding regions [J]. Archives of Virology, 2014, 159(5): 1149-1154.
[5] He Zhen, Yasaka R, Li Wenfeng, et al. Genetic structure of populations ofSugarcanestreakmosaicvirusin China: Comparison with the populations in India[J]. Virus Research, 2016, 211: 103-116.
[6] García-Arenal F, Fraile A, Malpica J M. Variability and genetic structure of plant virus populations [J]. Annual Review of Phytopathology, 2001, 39(1): 157-186.
[7] Gibbs A, Ohshima K.Potyvirusesand the digital revolution [J]. Annual Review of Phytopathology, 2010, 48(1): 205-223.[8] Viswanathan R, Balamuralikrishnan M, Karuppaiah R. Characterization and genetic diversity ofSugarcanestreakmosaicviruscausing mosaic in sugarcane [J]. Virus Genes, 2008, 36(3): 553-564.
[9] Bagyalakshmi K, Parameswari B, Chinnaraja C, et al. Genetic variability and potential recombination events in the HC-Pro gene ofSugarcanestreakmosaicvirus[J]. Archives of Virology, 2012, 157(7): 1371-1375.
[10]Fellers J P, Seifers D, Ryba-White M, et al. The complete genome sequence ofTriticummosaicvirus, a new wheat-infecting virus of the high plains [J]. Archives of Virology, 2009, 154(9): 1511-1515.
[11]Larkin M A, Blackshields G, Brown N P, et al. Clustal W and Clustal X version 2.0 [J].Bioinformatics,2007,23(21):2947-2948.
[12]Martin D P, Rybicki E P.RDP: detection of recombination amongst aligned sequences [J]. Bioinformatics, 2000, 16(6): 562-563.
[13]Martin D P, Lemey P, Lott M, et al. RDP3: a flexible and fast computer program for analyzing recombination [J]. Bioinformatics, 2010, 26(19): 2462-2463.
[14]Sawyer SA (1999) GENECONV: a computer package for the statistical detection of gene conversion [M]. Distributed by the author, Department of Mathematics, Washington University in Louis.
[15]Salminen M O, Carr J K, Burke D S, et al. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning [J]. AIDS Research and Human Retroviruses, 1995, 11(11): 1423-1425.
[16]Smith J M. Analyzing the mosaic structure of genes [J]. Journal of Molecular Evolution, 1992, 34(2): 126-129.
[17]Posada D, Crandall K A. Evaluation of methods for detecting recombination from DNA sequences: computer simulations [J]. Proceedings of the National Academy of Sciences, USA, 2001, 98(24): 13757-13762.
[18]Boni M F, Posada D, Feldman M W. An exact nonparametric method for inferring mosaic structure in sequence triplets [J]. Genetics, 2007, 176(2): 1035-1047.
[19]Gibbs M J, Armstrong J S, Gibbs A J. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences [J]. Bioinformatics, 2000, 16(7): 573-582.
[20]Ohshima K, Yamaguchi Y, Hirota R, et al. Molecular evolution ofTurnipmosaicvirus: evidence of host adaptation, genetic recombination and geographical spread[J]. Journal of General Virology, 2002, 83(6): 1511-1521.
[21]Tomitaka Y, Ohshima K. A phylogeographical study of theTurnipmosaicviruspopulation in East Asia reveals an “emergent” lineage in Japan [J].Molecular Ecology,2006,15(14):4437-4457.
[22]Nguyen H D, Tran H T N, Ohshima K. Genetic variation of theTurnipmosaicviruspopulation of Vietnam: a case study of founder, regional and local influences[J]. Virus Research, 2013, 171(1): 138-149.
[23]Guindon S, Dufayard J F, Lefort V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0[J]. Systems Biology, 2010, 59(3): 307-321.
[24]Tamura K, Stecher G, Peterson D, et al. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0[J]. Molecular Biology and Evolution, 2013, 30(12): 2725-2739.
[25]Huson D H, Bryant D. Application of phylogenetic networks in evolutionary studies [J]. Molecular Biology and Evolution, 2006, 23(2): 254-267.
[26]Posada D. jModelTest: phylogenetic model averaging [J]. Molecular Biology and Evolution, 2008, 25(7): 1253-1256.
[27]Page R D M. Tree view: An application to display phylogenetic trees on personal computers [J]. Bioinformatics, 1996, 12(4): 357-358.
[28]Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences [J]. Journal of Molecular Evolution, 1980, 16(2): 111-120.
[29]Dayhoff M O, Barker W C, Hunt L T. Establishing homologies in protein sequences [J]. Methods in Enzymology, 1983, 91: 524-545.
[30]Pamilo P, Bianchi N O.Evolution of theZfxandZfygenes: rates and interdependence between the genes [J]. Molecular Biology and Evolution, 1993, 10(2): 271-281.
[31]Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data [J]. Bioinformatics, 2009, 25(11): 1451-1452.
[32]Ogawa T, Tomitaka Y, Nakagawa A, et al. Genetic structure of a population ofPotatovirusYinducing potato tuber necrotic ringspot disease in Japan; comparison with North American and European populations [J]. Virus Research, 2008, 131(2): 199-212.
[33]Wei Taiyun, Yang Jinguang, Liao Fulong, et al. Genetic diversity and population structure of rice stripe virus in China [J]. Journal of General Virology, 2009, 90(4): 1025-1034.
[34]Yin Xiao, Zheng Fangqiang, Tang Wei, et al. Genetic structure of rice black-streaked dwarf virus populations in China [J]. Archives of Virology, 2013, 158(12): 2505-2515.
[35]Zhang Chengling, Gao Rui, Wang Jie, et al. Molecular variability ofTobaccoveinbandingmosaicviruspopulations [J]. Virus Research, 2011, 158(1): 188-198.
[36]Muhire B M, Varsani A, Martin D P.SDT: a virus classification tool based on pairwise sequence alignment and identity calculation [J]. PLoS ONE, 2014, 9(9): e108277.
[37]高芳銮, 沈建国, 史凤阳, 等. 中国马铃薯Y病毒的检测鉴定及CP基因的分子变异[J]. 中国农业科学, 2013, 46(15): 3125-3133.
[38]高芳銮, 沈建国, 史凤阳, 等. 马铃薯Y病毒pipo基因的分子变异及结构特征分析[J]. 遗传, 2013, 35(9): 1125-1134.
[39]Roossinck M J. Mechanisms of plant virus evolution [J]. Annual Review of Phytopathology, 1997, 35(1): 191-209.
(责任编辑:田 喆)
Molecular variation of HC-Pro gene ofSugarcanestreakmosaicvirus
He Zhen1,2, Li Wenfeng3, Zhang Zhixiang2, Li Shifang2
(1.SchoolofHorticultureandPlantProtection,YangzhouUniversity,Yangzhou225009,China; 2.StateKeyLaboratoryforBiologyofPlantDiseasesandInsectPests,InstituteofPlantProtection,ChineseAcademyofAgriculturalSciences,Beijing100193,China; 3.YunnanKeyLaboratoryofSugarcaneGeneticImprovement,SugarcaneResearchInstitute,YunnanAcademyofAgriculturalSciences,Kaiyuan661600,China)
The objective of this study is to assess the molecular evolution and divergence of SCSMV according to HC-Pro gene sequences. The HC-Pro gene sequences of SCSMV were obtained by RT-PCR, and analyzed by bioinformatic software, in aspect of recombination, phylogenetics, selection, demography, and gene flow. In the present study, 44 HC-Pro gene sequences were determined with a 70% lowest similarity; only one novel recombination site together with two previous reported sites were found in HC-Pro gene, suggesting that infrequent recombination events were distributed in this gene of SCSMV; one novel lineage (lineage Ⅲ) clustered by SCSMV sequences determined here was found; strong purifying selection was found in the HC-Pro gene of SCSMV. Our genetic study further indicates that the HC-Pro gene showed high genetic diversity in the genome of SCSMV.
Sugarcanestreakmosaicvirus; HC-Pro; molecular variation
2016-03-30
2016-04-23
植物病虫害生物学国家重点实验室开放课题(SKLOF201518);扬州大学科技创新培育基金(2015CXJ043)
S 435.661
A
10.3969/j.issn.0529-1542.2017.02.005
Research Reports
* 通信作者 E-mail: sfli@ippcaas.cn
猜你喜欢
当代水产(2022年7期)2022-09-20
科教新报(2020年45期)2020-03-15
读者·校园版(2019年20期)2019-10-18
小猕猴学习画刊(2019年8期)2019-09-16
宝藏(2019年3期)2019-03-28
广东第二课堂·小学(2019年1期)2019-03-06
宝藏(2019年2期)2019-01-14
少年漫画(艺术创想)(2018年7期)2018-11-19
艺术评论(2018年4期)2018-05-09
现代园艺(2018年3期)2018-02-10
