
N掺杂位置对四方相PbTiO3电子结构和光学性能的影响
2017-03-07 11:07:02李宏光闫金良
材料科学与工程学报 2017年1期
李宏光,闫金良
(1.鲁东大学 信息与电气工程学院,山东 烟台 264025; 2.鲁东大学 物理与光电工程学院,山东 烟台 264025)
N掺杂位置对四方相PbTiO3电子结构和光学性能的影响
李宏光1,闫金良2
(1.鲁东大学 信息与电气工程学院,山东 烟台 264025; 2.鲁东大学 物理与光电工程学院,山东 烟台 264025)
用第一性原理计算了N掺杂四方相钛酸铅的结构参数、形成焓、电子结构和光吸收,研究了N掺杂位置对钛酸铅性能的影响。N掺杂钛酸铅是典型的p型半导体,杂质能级主要由N 2p态贡献。N替位O(1)位置与N替位O(2)位置钛酸铅的形成焓差值很小。N替位O(1)位置钛酸铅在价带顶出现两条交互的杂质能级,N替位O(2)位置的钛酸铅在价带顶出现两条分离的杂质能级。N替位O(1)位置的钛酸铅的相对空穴数是1.729,N替位O(2)位置的钛酸铅相对空穴数为1.327。与N替位O(1)位置钛酸铅相比,N替位O(2)位置钛酸铅在300nm到1400nm区域光吸收强度明显增强。
钛酸铅; N掺杂; 电子结构; 第一性原理
1 引 言
透明导电氧化物(TCO)薄膜在太阳能电池、平板显示器、发光二极管、节能玻璃窗等领域得到广泛的应用。但迄今为止,工业上实际使用的TCO薄膜基本都是n型的。尽管过去在实验室里成功制备出多种P型TCO薄膜材料,但其性能还远远不能和n型TCO薄膜相比,体现在其电导率和空穴迁移率太低,性能稳定性很差。只有获得了性能合适的P型TCO薄膜材料,才能把TCO薄膜的半导体功能利用起来,将透明导电膜的应用领域从无源器件拓宽到有源器件。过渡族金属氧化物PbTiO3具有宽带隙和低空穴有效质量,是一种潜在的P型TCO材料[1]。PbTiO3在低温阶段呈四方对称性,属于P4mm对称群;在763K发生相变,PbTiO3呈立方体结构,属于Pm3m空间群[2]。在PbTiO3制备和退火过程中,杂质掺杂和缺陷能改变PbTiO3微观结构和原子之间的相互作用,调制PbTiO3的性能[3]。目前,多数研究集中在PbTiO3的O空位缺陷[4-5],而对于PbTiO3的P型掺杂关注很少。Ge F F用第一性原理计算了四方相PbTiO3的Pb空位、Ti空位和O空位的缺陷形成能和电子结构[6],带Pb空位和Ti空位的PbTiO3价带顶出现类受主能级,呈现P型导电性;带O空位的PbTiO3带隙中出现类施主能级,呈现n型导电性。本文探讨N替位掺杂四方相PbTiO3中不同位置O对PbTiO3的电子结构和光学性能的影响。
2 模型构建与计算方法
图1是四方相PbTiO3的晶体结构图,图示为8个单胞组成的2×2×2超晶胞,每一个单胞有5个原子。图中黑色球表示Pb原子,灰色球表示Ti原子,红色球表示O原子;明显看出,四方相PbTiO3晶胞中O原子有两种不同的位置,分别用O(1)和O(2)标注。在四方相PbTiO3超晶胞(2×2×2)中,用一个N原子分别取代O(1)和O(2),得到N掺杂四方相PbTiO3,相应的掺杂浓度为2.5at%。
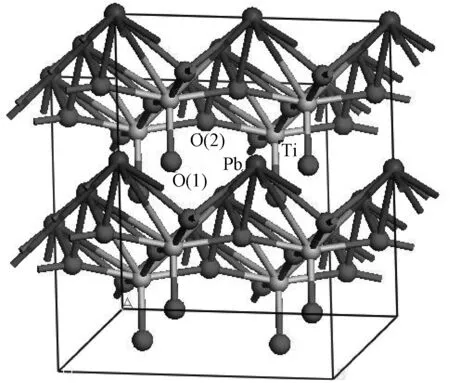
图1 本征四方相PbTiO3的晶体结构Fig.1 Crystal structure of intrinsic tetragonal PbTiO3
利用Materials Studio 5.0软件,在Castep模块下对四方相PbTiO3的晶体结构进行几何优化[7-8],优化质量为Ultra-fine,平面波的剪切能为380eV,计算
所使用的函数为GGA+PBE,每个原子受到的力小于0.01eV/Å,在布里渊区内对2×4×4 特殊k点进行积分求和;内应力小于0.02GPa,原子最大位移小于5.0×10-4Å,自洽收敛标准为5.0×10-7eV/atom,并选取Pb、Ti、O和N各原子的价电子组态分别为Pb-5d106s26p2, Ti-3s23p63d24s2, O-2s22p4,N-2s22p3。计算本征四方相PbTiO3和N掺杂四方相PbTiO3的形成焓、电子结构和光学性能。
3 计算结果与分析
3.1 N掺杂钛酸铅的结构参数和稳定性
表1列出本征四方相PbTiO3和N掺杂四方相PbTiO3的晶格常数与形成焓。对比本征四方相钛酸铅的晶格常量实验值和计算值可以看出,本征四方相钛酸铅的晶格常量实验值a=b=0.3880nm, c=0.4155nm;晶格常量计算值a=b=0.3843nm, c=0.4196nm;a、b计算值比实验值略小,误差为-0.95%;而晶格常量c值比实验值略大,误差为0.98%。我们的计算结果与实验结果吻合较好,印证了计算的合理性。N替位掺杂O(1)或O(2)位置后,晶格常量a和b变小,c变大。N替位掺杂O(2)位置后晶格常数沿c轴变化比较大,伸长量大约为0.0635nm,N掺杂O(2)位置钛酸铅原胞体积比本征钛酸铅增大了0.01nm3。这说明氮掺杂O(2)位置的钛酸铅要比氮掺杂O(1)位置的钛酸铅引起的晶格畸变大。计算得到本征四方相钛酸铅的形成焓值是-13.380eV,它是一个负值,表明本征钛酸铅是一种稳定结构。氮掺杂O(1)位置的钛酸铅的形成焓值为-12.731eV,氮替位掺杂O(2)位置的钛酸铅的形成焓为-12.739eV,氮掺杂后的晶体的形成焓值仍为负值,表明N掺杂钛酸铅晶体是热力学稳定的。
3.2 N掺杂钛酸铅的电荷差分密度
图2表示的是N替位掺杂两种不同的O位置时的电荷差分密度图,电荷差分密度可以用来分析晶体内原子之间的成键情况[10],在钛酸铅晶体里边,原子与原子之间成键的共价性或离子性可以解释电子的输运性质。从图中我们可以明显地看到,Ti 原子周围分布的电荷数目比较少,O(2)原子周围电荷呈球状均匀
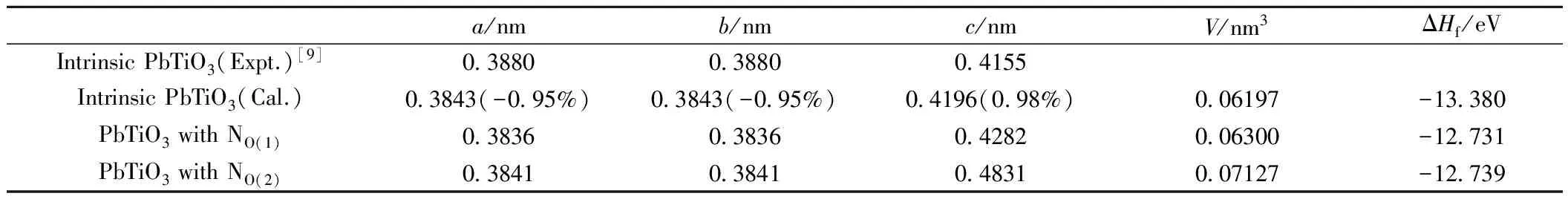
表1 本征与N掺杂四方相钛酸铅的晶格常数与形成焓

图2 N掺杂四方相钛酸铅的电荷差分密度图 (a) NO(1); (b) NO(2)。Fig.2 Electron density differences of N-doped tetragonal PbTiO3 (a) NO(1); (b) NO(2)
分布,这表明Ti 与O(2)之间具有典型的共价键的特性;虽然O(1)周围的电荷分布大体上也呈球状,但分布并不均匀,在靠近下边Ti位置的O(1)底部附近电荷聚集较多,而在Ti 与O(1)之间靠近Ti 位置的电荷分布明显减少,这是由于Ti 3d 态电子转移到O 2p,导致Ti 周围的电荷分布减少。因此Ti 与O(1)之间具有较强的离子键性能。当1个N 原子替位O(1)时,从差分电荷密度图可以看到Pb 周围具有均匀环状分布的、数量相对较少的电荷,因此Pb 与O(1)之间具有相对较弱的共价键特性。与O(1)-Ti 之间的化学键相比,N-Ti 之间既具有共价键特征,又具有离子键特征,但离子性明显增强。当一个N 原子替位O(2)时,与原来的O(2)位置差分电荷密度相比,发生了很大变化。在N 原子与Ti 原子之间靠近N 的位置分布数量较多的电荷,整体上N 原子周围的电荷呈球状分布,表明 Ti 原子与N 原子之间既有离子键的性能,又有共价键的性能。
3.3 能带结构和态密度
图3所示为本征四方相钛酸铅的能带结构和态密度,为了分析材料的半导体特征,图中只显示价带顶和导带底的能带分布,费米能级Ef设置在0eV能量位置。从图中我们发现能带结构图中价带顶与导带底位于布里渊区同一Gamma 点位置,表明四方相钛酸铅是典型的直接带隙半导体。计算得到的禁带宽度值是2.01eV,小于实验测量值3.6eV[11],这是由于密度泛函理论(DFT)是基于基态理论,它低估了激发电子态的交换关联势;然而,当我们对各种情况(本征和替位掺杂)设置一致的计算条件时,低估的带隙结果并不会对体系的电子结构和光学性质的分析造成影响。从总态密度和分态密度图中可以看出,价带顶主要由 Pb 6s态、O(1)和O(2)的2p 态构成;导带底主要由Ti 的3d态构成,我们计算得到的结果与前人的结果一致[2]。
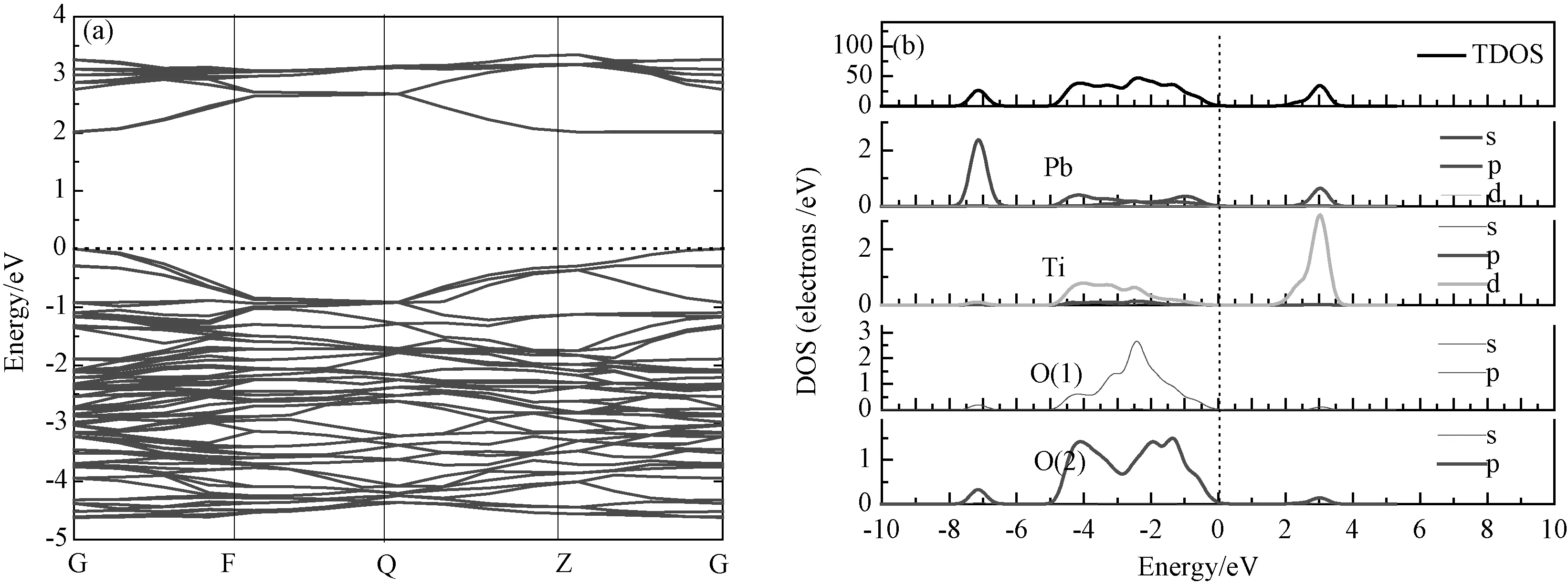
图3 本征钛酸铅的能带结构(a)和态密度(b)Fig.3 Band structures (a) and densities of states (b) of intrinsic PbTiO3

图4 N替位O(1)位置的钛酸铅的能带结构(a)和态密度(b)Fig.4 Band structures (a) and densities of states (b) of PbTiO3 with NO(1)
N替位O(1)位置钛酸铅的能带结构和态密度如图4所示,与本征钛酸铅的能带结构相比,N替位掺杂导致在价带顶出现两条交互在一起的杂质能级,价带顶和导带底的位置没有发生变化,但是由于杂质能级的出现,导致杂质能级以下的价带出现了下移的现象,移动了大约0.18eV,杂质能级最高能量点位于0.39eV。费米能级穿过杂质能级,费米能级以上出现空能态。N掺杂四方相钛酸铅表现出典型的p型半导体特性。从N替位O(1)位置钛酸铅的总态密度(TDOS)和分态密度(PDOS)可以看出,它的能带组成与本征的相似,价带顶主要由Pb 6s、O(1) 2p、O(2) 2p、N 2p态组成,导带底主要由Ti 3d组成,杂质能级由N 2p态贡献。从态密度图可以看出,费米能级以下属于电子占据态(全满),费米能级以上属于非占据态(空态)。通过对总态密度曲线从费米能级(0 eV)到价带顶这一部分曲线与横坐标所围成的面积积分,得到N替位O(1)位置的钛酸铅的相对空穴数是1.729[12]。
图5是N替位O(2)位置的钛酸铅的能带结构和态密度。N替位O(2)位置的钛酸铅的能带结构图中价带顶与导带底位于同一个 Gamma 点位置,N掺杂O(2)位置的钛酸铅是p型直接带隙半导体。与N掺杂O(1)位置的能带图相比,禁带宽度值更大一些(差值为0.05eV)。在费米能级之上有两条受主杂质能级,这两条杂质能级没有发生相互交叠现象。表明电子在价带内跃迁有两种可能性,一种是向能量较低的
受主态跃迁,另一种是向能量较高的受主态跃迁。从总态密度图可以看出,价带主要由3个峰构成,分别位于-7eV、-4eV、-2.25eV;导带仅有一个峰,位于3.25eV附近。位于-7eV的峰值主要由Pb 6s 态贡献,O(2) 2p态和N 2p态对它也有少量贡献;位于-4eV的峰主要由O(2) 2p态和Ti 3d态贡献;位于-2.25eV的峰主要由O(1)和O(2)的2p态贡献。处于导带3.25eV附近的峰由Pb 6p、Ti 3d态贡献。受主态主要由N 2p态贡献,O(1)对受主态没有贡献,O(2)对受主态有少量贡献。通过对总态密度价带部分(从0eV到价带顶区间)积分,得到相对空穴数为1.327。

图5 N替位O(2)位置的钛酸铅的能带结构(a)和态密度(b)Fig.5 Band structures (a) and densities of states (b) of PbTiO3 with NO(2)
3.4 光学吸收
半导体材料的光学吸收主要归因于半导体内电子的跃迁过程,当光源照射到晶体材料表面时,由于晶体内电子吸收了一定频率的光子的能量,使电子从低能状态激发到高能状态;同时,晶体当中的晶格振动也可以和入射光波发生相互作用,从而发生声子的光吸收与电子跃迁过程。光学性质的研究是探究半导体光电器件物理性质的重要方面之一,它主要涉及到光波辐射与材料之间的相互作用,而半导体的光学性质又与它的能带结构、态密度相联系,研究半导体材料的光学性质,可为拓宽它的应用领域打下基础。
图6所示为本征钛酸铅和N替位O(1)、O(2)位置的钛酸铅的光吸收图谱。为了使我们计算得到的光学性质与实验值吻合,我们在分析时作了剪刀算符修正[13],修正值 ΔE=1.59eV。从图中可以看出,在短光波长区域(0~100nm),光吸收曲线的振荡幅度比较大,短光波长对应着能量比较高的光子。在大约150nm波长处,出现一个最高吸收峰(330000cm-1),这个吸收峰可能来自于从价带的占据态(O 2p)到导带的非占据态(Pb 3p和Ti 3d)之间的带间跃迁。从图中我们发现,在光波长大于150nm 以后,光吸收的值呈线性减小的规律,光吸收边截止于大约350nm 附近。我们知道,光的波长与光子能量之间的关系可用公式E=hν=h c/λ表达,其中,E代表光波的光子能量,h 代表普朗克常量(大约等于4.1356674335×10-15eV·s),ν 代表光波频率,c 代表光在真空中的传播速度,λ代表光的波长。通过这个公式可以进行光子能量与光波长之间的换算。通过计算得知,吸收边350nm 对应于3.54eV, 这表明电子是从价带内跃迁到导带的。在150nm附近,本征四方相钛酸铅的峰值最大,N替位O(2)位置次之,N替位O(1)位置峰值最小。这表明钛酸铅掺入N之后,在150nm波长附近的光吸收强度变弱。在可见光和近红外波长区域,N掺杂钛酸铅的光吸收强度明显大于本征钛酸铅的光吸收强度。与N替位O(1)位置钛酸铅光吸收曲线相比,N替位O(2)位置钛酸铅在300nm到1400nm区域光吸收强度大大增加,这说明N替位O(2)位置钛酸铅所具有的潜在的光催化能力要比N替位O(1)位置钛酸铅好,而N替位O(1)位置钛酸铅是一种潜在的P型透明导电氧化物。
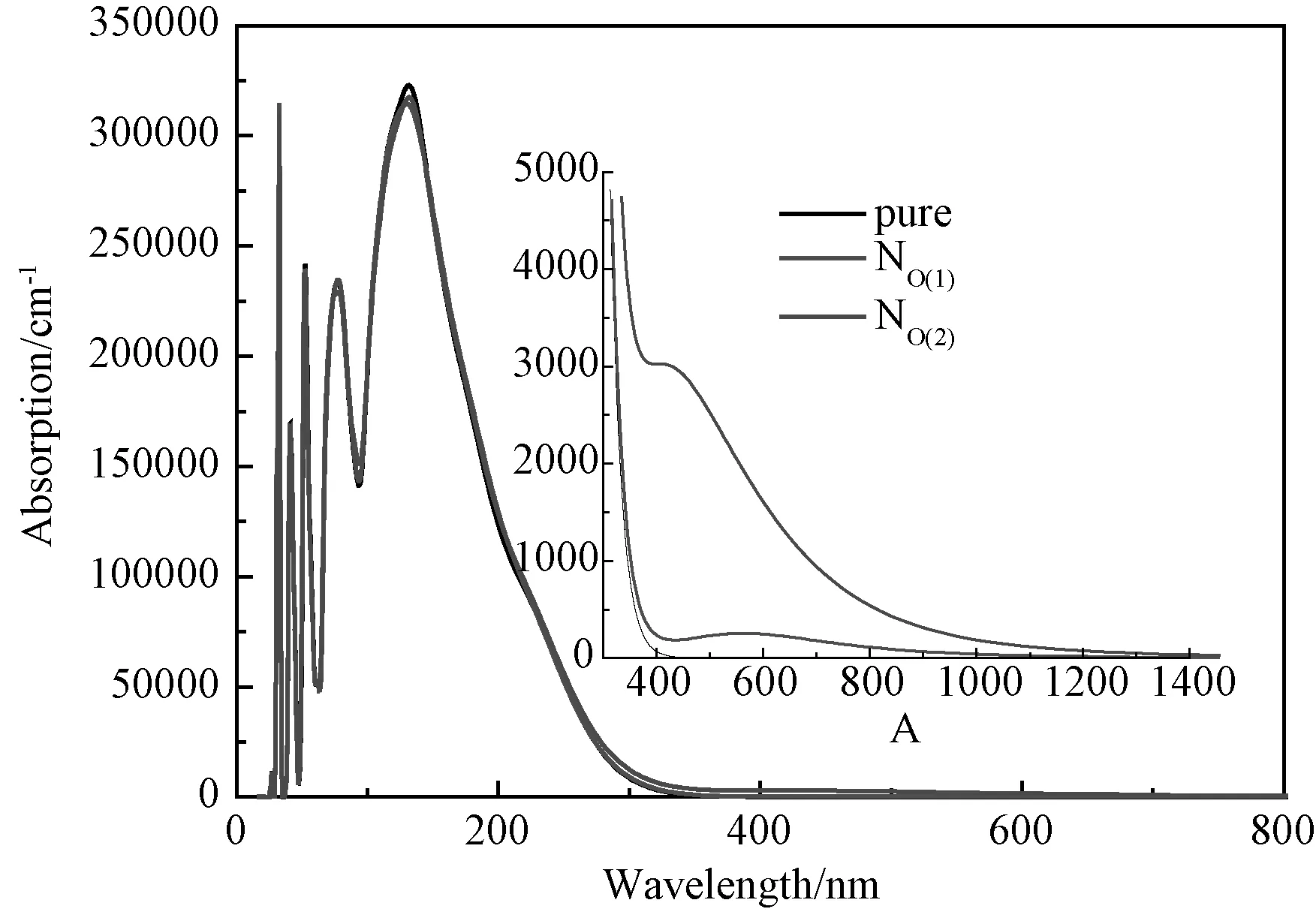
图6 本征钛酸铅和N掺杂钛酸铅的光吸收图谱Fig.6 Absorption of intrinsic PbTiO3 and N-doped PbTiO3
研究了N掺杂四方相钛酸铅的能带结构、态密度和光学吸收性质。N掺杂后钛酸铅的晶胞体积膨胀,掺N体系的形成焓值变大,N替位O(1)位置与N替位O(2)位置钛酸铅的形成焓差值很小。N替位O(1)位置钛酸铅在价带顶出现两条交互的杂质能级,N替位O(2)位置的钛酸铅在价带顶出现两条分离的杂质能级。N掺杂钛酸铅是典型的p型半导体,杂质能级主要由N 2p态贡献。N替位O(1)位置钛酸铅的相对空穴数是1.729,N替位O(2)位置的钛酸铅相对空穴数为1.327。与N替位O(1)位置钛酸铅相比,N替位O(2)位置钛酸铅在300nm到1400nm区域光吸收强度大大增加。N替位O(2)位置钛酸铅是潜在的光催化材料,而N替位O(1)位置钛酸铅是一种潜在的P型透明导电氧化物。
[1] Kim T W, Lee D U. Structural and optical properties of PbTiO3films grown on indium tin oxide-coated glass by metal organic chemical vapor continuous cooling process [J]. Solid State Communications, 1995, 96(2): 95~99.
[2] Hosseini S M, Movlarooy T, Kompany A. First-principle calculations of the cohesive energy and the electronic properties of PbTiO3[J]. Physica B, 2007, 391(2): 316~321.
[3] Hashima H, Suzuki Y, Ogawa S, Nakajima S. Preparation of PbTiO3thin films by ion-beam sputtering [J]. Thin solid films, 1996, 281(1~2): 463~465.
[4] Zhang Z, Wu P, Lu L. Study on vacancy formation in ferroelectric PbTiO3from ab initio[J]. Applied Physics Letters, 2006, 88(14): 142902.
[5] 孟亭,赵辉.O空位对PbTiO3晶体影响的第一性原理研究[J]. 天津师范大学学报(自然科学版), 2011, 31(2): 46~50.
[6] Ge F F, Wu W D, Wang X M, et al. The first-principle calculation of structures and defect energies in tetragonal PbTiO3[J]. Physica B, 2009, 404(20): 3814~3818.
[7] 李毅杰,田汉民,刘剑飞.Al掺杂锐钛矿型二氧化钛的第一性原理研究[J]. 材料科学与工程学报, 2013, 31(2): 305~309.
[8] 赵银女.氧空位对Ga1.5In0.5O3透明导电氧化物性能的影响[J]. 材料科学与工程学报, 2015, 33(3): 396~400.
[9] Shimada T, Ueda T, Wang J, Kitamura T. Hybrid Hartree-Fock density functional study of charged point defects in ferroelectric PbTiO3[J]. Physical Review B, 2013, 87(17): 174111.
[10] 赵银女.Sn掺杂 Ga1.375In0.625O3透明导电氧化物的第一性原理计算[J]. 材料科学与工程学报, 2015, 33(1): 46~50.
[11] Yadav H O. Optical and electrical properties of sol-gel derived thin films of PbTiO3[J]. Ceramics International, 2004, 30(7): 1493~1498.
[12] Wu H C, Peng Y C, Chen C C. Effects of Ga concentration on electronic and optical properties of Ga-doped ZnO from first principles calculations[J]. Optical Materials, 2013, 35(3): 509~515.
[13] Brik M G, Sildos I, Kiisk V. First-principles calculations of optical and electronic properties of pure and Sm3+-doped TiO2[J]. Physica B, 2010, 405(10): 2450~2456.
Electronic Structures and Optical Properties of N-doped Tetragonal PbTiO3with Different Doping Sites
LI Hongguang1, YAN Jinliang2
(1.School of Information and Electrical Engineering, Ludong University, Yantai 264025, China; 2.School of Physics and Optoelectronic Engineering, Ludong University, Yantai 264025, China)
Structural parameters, formation enthalpies, electronic structures and optical absorption of N-doped tetragonal PbTiO3were calculated by using first-principles. Effect of N-doping sites on the properties of PbTiO3was analyzed. The N-doped PbTiO3is a typical P-type semiconductor, the acceptor energy levels of which are composed mainly of N 2p states. Formation enthalpies of N-doped PbTiO3with NO(1)and NO(2)are almost identical. There are two overlap levels above the valence band of PbTiO3with NO(1), whereas two segregated acceptor levels appear above the valence band of PbTiO3with NO(2). The relative hole numbers are 1.729 and 1.327 for N-doped PbTiO3with NO(1)and NO(2), respectively. Compared with PbTiO3with NO(1), PbTiO3with NO(2)displays a strong absorption in the wavelength of 300nm-1400nm.
PbTiO3; N-doping; electronic structure; first-principles
1673-2812(2017)01-0014-04
2015-11-20;
2016-02-25
国家自然科学基金资助项目(11504155)和山东省自然科学基金资助项目(ZR2016FM38)
李宏光(1964-),男,副教授,主要研究方向为光电薄膜与器件。E-mail: ldu_LHG@sina.com。
闫金良(1965-),男,教授,主要研究方向为光电薄膜与器件。 E-mail: yanjinliang8@sina.com。
TN304.2; O474
A
10.14136/j.cnki.issn 1673-2812.2017.01.004
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
农业工程学报(2022年7期)2022-07-09 07:06:56
数学物理学报(2020年1期)2020-04-21 06:00:22
数学物理学报(2018年4期)2018-09-14 03:40:58
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
Chinese Journal of Chemical Engineering(2017年5期)2017-05-28 10:22:54
功能材料(2016年1期)2016-05-17 03:38:24
材料科学与工程学报(2016年5期)2016-02-27 07:11:28
中南民族大学学报(自然科学版)(2011年1期)2011-08-23 09:14:38
物理学报(2010年6期)2010-09-08 06:05:24
