
用静电势电荷预测达格列净衍生物溶解度和光谱
2017-02-01 01:46吴淑曼洪琴彭自珍吴林钢王玉娜濮亚敏胡俊逸钟爱国
当代化工 2017年12期
吴淑曼,洪琴,彭自珍,吴林钢,王玉娜,濮亚敏,胡俊逸, 钟爱国
用静电势电荷预测达格列净衍生物溶解度和光谱
吴淑曼,洪琴,彭自珍,吴林钢,王玉娜,濮亚敏,胡俊逸, 钟爱国*
(台州学院医药化工学院, 浙江 台州 318000)
基于密度泛函理论(DFT)和密度泛函活性理论(DFRT)和方法(MO6-2X/6-31+G**),利用软件Chemoffice 2004分子建模,用Gaussion 09W软件优化分子结构,用ACD Lab 6.0 软件预测新型降糖药达格列净分子及其衍生物分子的水溶液溶解度参数等,优化了16种降糖药达格列净及其衍生物的结构,得到了桥头O原子的密立根电荷(Mul-O),桥头O原子的自然原子轨道电荷(NBO-O),桥头O原子的静电势电荷(ESP-O)和桥头O原子的海秀巴赫电荷(HIR-O)。探讨了常见的10类基组和泛函对ESP 电荷值的影响。对比分析其它种类电荷数据发现,桥氧原子的ESP-O电荷与达格列分子及其衍生物水溶液的分子溶解度存在较强烈的线型关系(=3.699+24.66,=0.929)。基于密度泛函理论与方法(DFT B3LYP/sto-3 g),对比展示了药达格列净及其单取代衍生物的三大分子吸收光谱(红外光谱,拉曼光谱和紫外吸收光谱)的差异,并表征了它们的峰位和峰型。
密度泛函理论;密度泛函活性理论;达格列净;原子电荷;分子光谱
达格列净(Dapagliflozin)(图1),是美国Bristol-Myers Squibb公司和瑞典Astra Zeneca公司联合开发了钠-葡萄糖协同转运蛋白2抑制剂(SGLT2),该药物分别在2012年和2014年先后在欧洲药品管理局和美国FDA批准上市[1]。达格列净通过特分子能特异性地抑制SGLT-2的活性来达到减小肾小管葡萄糖重吸收的目的,增加葡萄糖在尿液中放入排泄,从而起到降血糖的作用。其特殊作用机制,在于不会出现低血糖、恶心、水肿等不良反应,使达格列净称为的新型口服降糖药。正是因为达格列净抑制剂的成功,人们纷纷加入到对其的研究开发。甚至有些公司为了寻找更大的专利空间,转向SGLT-2 家族这个新型靶点的研究开发和结构改造上[2,,3]。然而在实际开发过程中由于受到其他条件因素的制约,很难确定药物的一些基本的物化性质,其中就包括分子物质的溶解度(log)、脂水分配系数(log)和分子酸碱度(pab)等。相较于脂水分配系数(log)和分子酸碱度(pa,b),分子溶解度的预测更是一个困难的突破点。这是因为物质本身溶解过程是很复杂的一个物理化学相变化过程。不仅需要考虑溶质分子之间的相互作用力,还需要衡量相变过程。鉴于先前已有研究采用密度泛函理论(DFT)所得的原子电荷数据来预测药物分子及其衍生物的pb,并且得到了良好的关于药物pb预测的线性方程[4-7]。因此本文将采用密度泛函理论的方法,在DFT MO6-2/6-31+G(d,p)水平下优化达格列净及其衍生物的分子结构,通过计算得到相关性最高的关于预测溶解度的线性方程,从而起到预测达格列净其他衍生物的溶解度的作用。这一方法对于研究改造达格列净及其衍生物的药理作用及其毒副作用有着极其重要的意义。此外本文还采用密度泛函活性理论(DFRT)分析了达格列净一元取代及多元取代分子的红外吸收(IR)、拉曼和紫外光谱(UV-Vis),分析和验证了取代基对达格列净整体结构的影响,明确其稳定的空间结构。
1 实验部分
1.1 分子建模
达格列净的结构如图1所示,通过改变椅式结构上的取代基以及其位置获得其衍生物(图1),来研究其4种常见原子电荷:密立根电荷(Mulliken)、静电势电荷(ESP)、自然原子轨道电荷(NBO)和海秀巴赫电荷(HIR)变化与采用ACD Lab 6.0 软件测得的水溶液溶解度对数log的线性关系。本文再用了−F、−Cl、−CH3作为取代基在糖苷环R1、R2、R3、R4上进行一元取代、二元取代、三元取代和四元取代,总共得到16种达格列净的衍生物。
1.2 计算方法
使用 Chem 3D 和 Chem Draw 8.0 软件,构建16个格列净衍生物的模型,并使其能量最低化;在GaussView 5.0.9 软件中,使用 DFT/MO6-2X 方法在6-31+G(d,p)水平下优化结构,计算关键词为M06-2x,iop(5/33)=1,pop=(nbo,chelpg,hirshfeld)的条件下,分别计算16个达格列净及其衍生物的量化描述符参数。记录糖苷环上桥头氧原子的Mulliken、ESP、NBO 和HIR 的电荷值;运用ACD Labs 6.0软件,得到16种达格列净及其衍生物相应的水溶液的溶解度(log)预测值;分别将16种达格列净衍生物的Mulliken、ESP、NBO 和HIR 的电荷值得到的溶解度(log),使用Origin 8.5软件处理数据,最后确定相关系数(值)最大的线性回归方为预测方程。红外光谱(IR)和拉曼光谱在DFT/B3LYP/STO-3G水平优化结构,采用 Frequency 方法分别计算得到16种达格列净衍生物的频率。计算由Gaussian 09W 完成。谱图处理由 Origin 8.5 完成。紫外吸收光谱在TD DFT/B3LYP/3-21G水平分别模拟计算得到16种达格列净衍生物的频率,谱图处理由 Origin 8.5 完成。
2 结果与讨论
2.1 常见基组对原子电荷的影响
骨架原子电荷是对分子体系中点电荷分布最简单、最直观的描述方式, 在实际和理论应用中都有着重要的意义。自从1955年密立根(Mulliken)原子电荷被提出以来, 至今已有60余种原子电荷及其计算方法被提出, 近年还有众多研究者在改进方法。这些方法如 Mulliken电荷、分子中的原子(AIM)电荷、Hirshfeld电荷、Hirshfeld电荷、Hirshfeld布居(ADCH)电荷、自然布居分析(NBO)电荷、Merz-Kollmann(MK)电荷、分子中的原子(AIM)电荷、Merck 分子力场 94 (MMFF94)电荷、AM1-BCC电荷、Gasteiger电荷、电荷模型 2(CM2)电荷以及电荷均衡(QEq)方法电荷等12种常见原子电荷[8]。我们采用5种重要的原子电荷计算方法, 从不同角度比较了它们的优缺点。首先检验常见的10种不同基组计算的5种达格列净小分子的桥头氧原子电荷值, 表1列出了10组密度泛函基组对5种常见电荷的计算结果。可以看出不同方法得到的桥头氧原子电荷的数值大小会有所不一样。Hirshfeld电荷整体偏小,许多氧原子电荷的绝对值大小都不足 0.7。ADCH 是对 Hirshfeld 方法的校正, 校正后氧原子电荷绝对值普遍增大,桥氧原子电荷绝对值比校正前均大于0.8以上。对于由碳、氢、氯、氧构成的中性有机分子达格列净,Mulliken、ACDH、ESP和NBO电荷在数量级上比较接近。基组从3-21G提升至6-311+G(d,p)的过程中,各种基组方法计算的结果都有不同程度的变化, 通常被认为具有很好基组稳定性的 NBO 电荷的变化值甚至大于公认基组稳定性差的 Mulliken电荷。采用10种不同的基组来检测达格列净的6号桥头氧原子的Mulliken、HIR、NBO、ESP和ACDH电荷受基组的依赖程度。由结果可知,基组对达格列净的6号桥氧原子的的5种电荷的影响不尽相同。其中Mulliken值的相对标准偏差STD=±7.6%,受基组影响较大,依赖性较高;而NBO、ACDH、ESP和NBO电荷值的相对标准偏差STD≤±1.5%,受基组影响较小,依赖性较小。
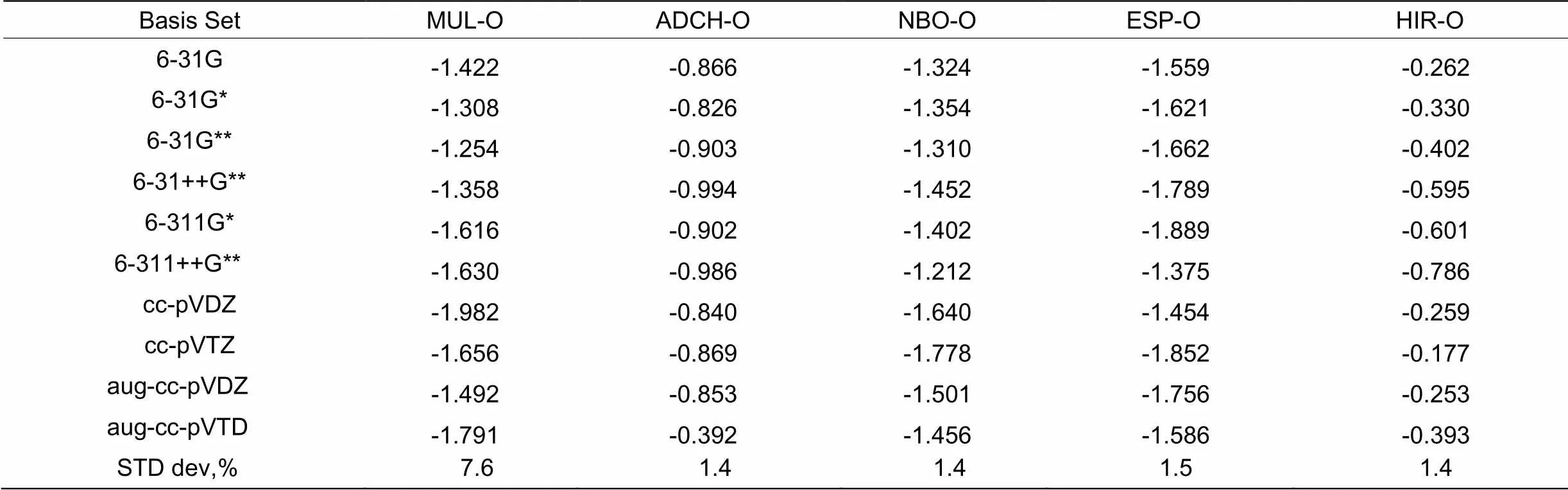
表 1 常见基组对达格列净6号桥氧原子电荷影响
2.2 常见泛函对原子电荷的影响
表2比较了10种常见的密度泛函方法计算的达格列净的6号桥氧原子的Mulliken、HIR、NBO、ESP和ACDH电荷对泛函的依赖程度。统一采用6-31+g(d,p) 基组的前提下,筛选并采用了包括BLYP、B3LYP、TPSS和X3LYP等在内的10种泛函得出了达格列净的6号桥氧原子的5种电荷值对泛函的依赖程度。由表2可知,达格列净的6号桥氧原子的Mulliken值的相对标准偏差STD=±2.0%,受泛函影响较大,依赖性大;而NBO、ESP和NBO电荷值的相对标准偏差STD≤±1.6%,受基组影响较小,依赖性较大。结合分析表1和表2数据可以看出,用DFT电荷分解方法,采用不同的基组和泛函组合,Mulliken值对基组和泛函依赖性大,而HIR、NBO、ESP和ACDH电荷值没有明显的依赖性,这一计算结果与实验值相符合[5,6]。
2.3 达格列净及其衍生物的溶解度
由DFT 计算所得的16种达格列净的衍生物桥头O的Mulligen、ACDH、ESP、NBO 和HIR 的电荷值以及由 ACD Labs 6.0 软件预测得到的相应衍生物水溶液的溶解度log(如表2所示)。为直观的表现电荷值变化与log值关系,分别用桥头O原子的Mulligen、ACDH、ESP、NBO 和HIR 的电荷值与 log值作图。Mull-O 电荷对 log相关系数较小(=0.434);ESP-O 对 log如图3所示,相关系数(=0.929,SD=0.166);NBO-O 电荷与 log之间(=0.114,SD=0.440);HIR-O电荷对log之间=0.520。5个线性方程中,ESP-6O电荷值与log值的线性方程的值最大(0.929),即相关性最好。
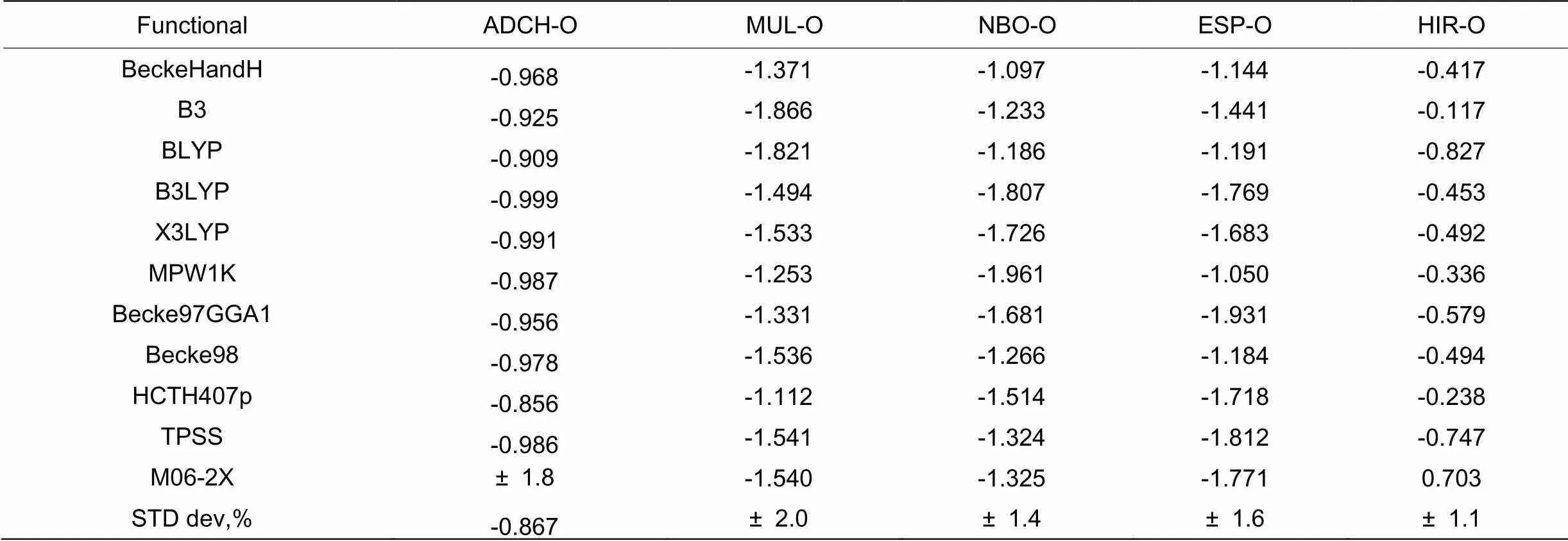
表 2 常见泛函对达格列净6号桥氧原子电荷影响
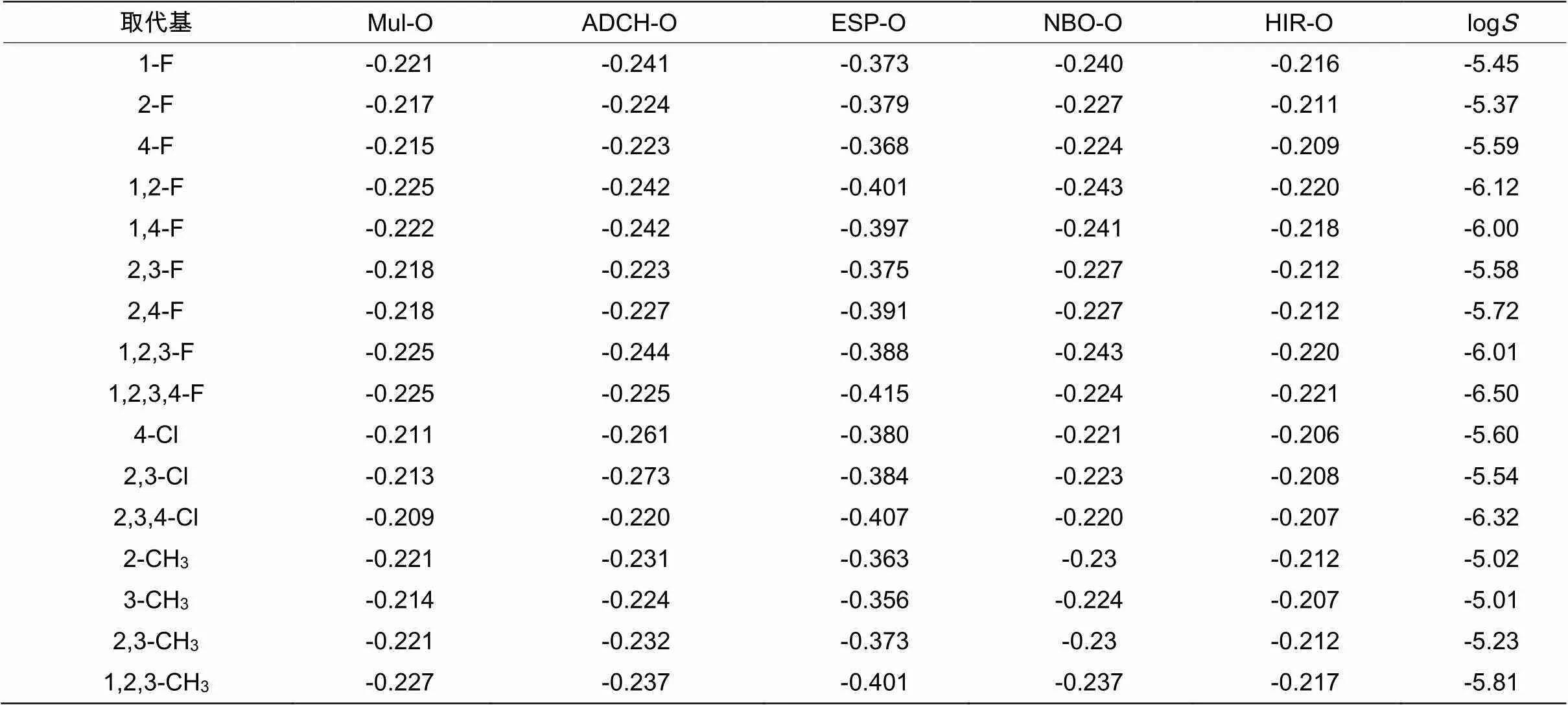
表3 16种达格列净衍生物6号桥氧原子5种电荷计算
3 达格列净及其衍生物的分子光谱
3.1 红外吸收光谱
分子的IR光谱是分子骨架振动-转动属性的内在反应。在DFT/B3LYP/STO-3G水平优化结构,图3展示了2-F取代的达格列净衍生物的吸收峰:3 330 cm-1处的吸收峰为苯环上C-H的伸缩振动;1 656、1 593、1 467 cm-1处的吸收峰为苯环骨架的伸缩振动;1 341 cm-1处的吸收峰为饱和C-H的弯曲振动;1 251 cm-1处的吸收峰为芳族的 =C-O-C的反对称伸缩振动;1 143 cm-1处的吸收峰为C-O-C的对称伸缩振动。伯醇的伸缩振动的吸收峰和C-Cl的对称伸缩振动的吸收峰和C-F的反对称伸缩振动均有出现,但较弱。

图2 ESP-6O vs logS值的线性关系
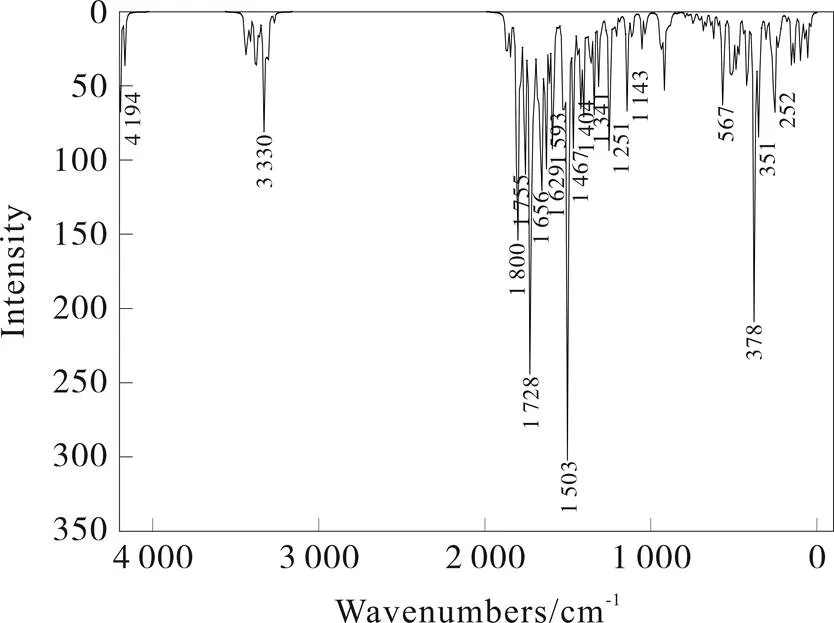
图3 2-氟取代的达格列净衍生物红外光谱图
3.2 拉曼吸收光谱
在DFT/B3LYP/STO-3G水平优化结构,得到达格列净拉曼光谱图(图4)。

图4 达格列净拉曼吸收光谱图
在4 162 cm-1处的吸收峰为OH的对称伸缩振动;在3 384 cm-1处的吸收峰为苯衍生物的C-H对称伸缩振动;在3 330 cm-1处的吸收峰为C-H的对称伸缩振动;在3 267 cm-1处的吸收峰为C-H的反对称伸缩振动;在1 809 cm-1处的吸收峰为C-H的弯曲伸缩振动;在945 cm-1处的吸收峰为伯氯的伸缩振动峰。一元取代(2-F)的达格列净衍生物拉曼光谱图基本相似(省去)。
3.3 紫外吸收光谱
在DFT/B3LYP/3-21G水平优化结构,分别计算得到达格列净紫外光谱图,显示了达格列净衍生物在282.7 nm处有最大吸收波长,位于近紫外区。这是由于π→π*跃迁,吸收波长较长。2-F 取代的达格列净衍生物在281.3 nm处有最大吸收波长,位于近紫外区;2-CH3取代的达格列净衍生物在186.9 nm处有最大吸收波长,位于近紫外区。
4 结论
(1)基于DFT理论电荷分解法,检测并分析达格列净的6号桥氧原子的Mulliken、HIR、NBO、ESP和ACDH电荷对10种基组和泛函的依赖程度。其中,Mulliken值对基组和泛函依赖性大,而HIR、NBO、ESP和ACDH电荷值对基组和泛函没有明显的依赖性。
(2)采用密度泛函活性理论与方法,在DFT MO6-2X泛函和6-31+G(d,p)基组下,优化了16种达格列净及其衍生物的分子结构,可知6号桥头氧原子的ESP电荷与log值的相关性最好,相关系数达0.929,最大偏差低于2%;
(3)在B3YLYP/sto-3g水平下,对达格列净衍生物进行红外、紫外和拉曼光谱的分析,其基本结构较稳定,取代基及位置不同未对其骨架结构造成特别大的影响。
[1]刘文杰,陈林,石克金,等.达格列净的合成[J].化学研究与应用,2016,28(4):530-533.
[2]陈燕梅,李志勇.达格列净的临床应用进展[J].重庆医学,2013,42(34):4214-4216.
[3]巫凤娟,杨臻峥,孙大柠.降糖药Dapaglinozin[J].药学进展.2009,33(7):334-336.
[4]万惠新,沈竞康.2型糖尿病治疗靶点钠-葡萄糖共转运蛋白2抑制剂研究进展.药学学报,2012:47(6):716-724.
[5]裴诗恩,黄颖琦,乔骏驰,等. 基于静电势电荷的抗击埃博拉病毒新药物法匹拉韦及其衍生物水溶解度log值预测[J].当代化工,2017,46(1):31-34.
[6]李余,裴诗恩,钟爱国,等.抗埃博拉病毒药物法匹拉韦及其衍生物pb值预测[J].当代化工,2016,45(7):1503-1504
[7]陈红,丁俊杰,丁晓琴. 从头计算研究水溶液中有机化合物的 pa值[J].计算机与应用化学,2007,24(5):580-582.
[8]卢 天, 陈飞武.原子电荷计算方法的对比[J].物理化学学报,2012, 28 (1): 1-18.
Prediction of the Solubility and Molecular Spectra of New Antidiabetic Drug Dapagliflozin and Its Derivatives by Density Functional Theory and Electrostatic Potential Charge
WU Shu-man, HONG Qin, PENG Zi-zhen, WU Lin-gang, WANG Yu-na, PU Ya-min, HU Jun-yi, ZHONG Ai-guo
(School of Medicine and Chemical Engineering, Taizhou University, Zhejiang Taizhou 318000, China)
Based on the density functional theory (DFT) and time-dependent density functional theory (DFRT) and method (MO6-2X/6-311G), the software of Chemoffice 2004 was used to establish molecular model, Gaussion 09W software was used to optimize the molecular structure, ACD Lab 6 software was used to predict the aqueous solubility parameters of new type antihyperglycemic farxiga molecule and its derivatives, molecular structures of 16 kinds of hypoglycemic drugs farxiga and its derivatives were optimized, the O atom charge Milliken bridge (Mul-O) was obtained as well as the natural atomic orbital charge bridge O atom (NBO-O), the electrostatic potential charge bridge O atom (ESP-O) and bridge O atom haixiu Bach charge (HIR-O). The influence of 10 kinds of basic groups and functional on the ESP charge value was discussed. The results show that there is a strong linear relationship between the ESP-O charge of the bridging oxygen atom and the solubility of the aqueous solution of the molecule and its derivatives (=3.699+24.66,=0.929). The differences of the absorption spectra of the four molecules and their peaks were compared and analyzed.
density functional theory; density functional activity theory; dapagliflozin; atomic charge
TQ 016
A
1671-0460(2017)12-2412-04
浙江省自然科学基金(LY15B030001)和浙江省大学生创新训练计划项目(201710350015)资助。
2017-04-16
吴淑曼(1994-),女,浙江温州人,研究方向: 化学工程与工艺专业。E-mail:915926977@qq.com。
钟爱国(1964-),男,教授,硕士,研究方向:计算化学。E-mail:zhongaiguo@tzc.edu.cn。
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
童话王国·文学大师班(2022年2期)2022-02-05
昆明医科大学学报(2021年12期)2021-12-30
椰城(2021年12期)2021-12-10
少儿科学周刊·少年版(2021年22期)2021-01-17
火工品(2019年6期)2019-06-05
厦门大学学报(自然科学版)(2018年2期)2018-04-11
物理学报(2017年21期)2017-11-10
腐蚀与防护(2016年7期)2016-09-14
当代化工研究(2016年5期)2016-03-20
