
环氧树脂固化过程中的黏弹性分析
2017-01-03 01:59李润明石家华
化学研究 2016年6期
李润明,田 培,石家华
(河南大学 化学化工学院,河南 开封 475004)
环氧树脂固化过程中的黏弹性分析
李润明,田 培,石家华*
(河南大学 化学化工学院,河南 开封 475004)
环氧树脂在电子线路板、航空工业以及高性能运动器材行业有着广泛的应用,其成型工艺普遍采用反应注射浇铸成型技术,注射温度及时间、保温温度及时间对得到尺寸满意的制品具有重要意义.通过升温测试,获得了树脂黏度和动态模量对温度的演变关系;通过一系列不同温度的保温测试,获得了凝胶化时间对温度的关系.结果表明,可以通过黏弹性能有效示踪固化过程中的结构演变,从而为反应成型加工提供有效、有用的工艺参数.
环氧树脂;固化;复数黏度;动态模量
环氧树脂固化后得到的高聚物具有如模量高、变形收缩率小、制品尺寸稳定等优良的力学性能,且具备对碱及大部分溶剂稳定等良好的化学抗蚀性,同时固化树脂对金属和非金属材料的表面具有优异的粘接强度,且其介电性能良好,因而被广泛应用于国防以及国民经济的各部门,其中用于电子行业制作印刷线路板的数量最大[1-3].
由于环氧树脂固化后不溶、不熔,因此印刷线路板成型工艺只能采取反应注射浇铸成型,即树脂首先需要通过流动充满模腔,然后再固化形成最终具备一定形状的致密体[4].充模过程中的主要影响因素是树脂的流动性,即树脂的黏度,其主要取决于树脂配方及模腔温度及充模时间.成型阶段除了黏度增加导致树脂丧失流动性外,树脂模量也开始快速增加直至最终完全固化,该过程主要的影响因素是体系的热历程如保温温度及保温时间等[4].该行业目前仍普遍采用流动黏度法获取相应的工艺参数[5-6],但由于固化会导致黏度较快升高甚至丧失流动性,很难准确、完整获取树脂加工所需的工艺参数.本文旨在通过跟踪固化过程中的黏弹性能变化来寻找合适的注射温度区间和固化保温温度及时间.
1 实验部分
1.1 材料
可固化型环氧树脂预浸料,牌号为A2049,昆山鹏博电子材料有限公司.
1.2 仪器及测试
旋转流变仪为TA Instrument Discovery HR-2,温控系统为环境控制炉,测试夹具为25 mm可抛弃平行板.
小振幅振荡升温测试:分别以2、3、4、5 ℃/min的升温速率从35 ℃升温至190 ℃,振荡频率为1 Hz,振荡幅度控制在树脂的线性黏弹区内.
恒温小振幅振荡:分别在110、115、120、130和140 ℃保温执行小振幅振荡1 h,振荡频率1 Hz,振荡幅度控制在树脂的线性黏弹区内.
2 结果与讨论
2.1 升温固化过程中的复数黏度及动态模量演进

图1 升温过程中复数黏度(a)和动态模量(b)演变
可固化环氧树脂在升温过程中的黏度演变如图1a所示,可以看出,升温过程中树脂的动态模量演变可粗略分为4个阶段:
Ⅰ) 35~75 ℃,黏度随温度单调降低,对于无交联低聚物而言,随着温度的升高分子链活动性增强,从而导致其流动性增加,体系黏度减小;
Ⅱ) 75~125 ℃,黏度随温度继续单调降低,但降低趋势有所减缓;
Ⅲ) 125~150 ℃,黏度随温度升高急剧,表明固化反应在该温度区间进行很快;
Ⅳ) 150 ℃以上,黏度随温度升高增加趋缓,表明固化反应接近完全.
从图1a可以看出,树脂注射充模温度应该控制在最低黏度对应的温度之下,此时,体系的黏度相对较低,可以保证树脂有较好的流动性.
对于低聚物无交联反应发生的情况而言,随着温度升高,链的活动能力增强,必然导致弹性模量G′和黏性模量G″均相应降低.一旦交联反应显著发生,体系的动态模量势必增加.可固化型环氧树脂升温过程中的动态模量演变如图1b所示,可以看出,升温过程中树脂的黏度演变可粗略分为5个阶段:
Ⅰ) 35~75 ℃,动态模量随温度单调降低,且G″>G′,表明此过程中的树脂实际上处于流体熔融状态;
Ⅱ) 75~125 ℃,动态模量随温度继续单调降低但降低幅度趋缓,且G″>G′,此过程中的树脂仍处于流体熔融状态;
Ⅲ) 125~138 ℃(138 ℃为凝胶化温度[7]),动态模量随温度快速升高,但G″>G′,表明树脂开始快速反应,但仍具备流动能力;
Ⅳ) 138~150 ℃,动态模量随温度继续快速升高,但G′开始超过G″,树脂流动能力丧失,转变为凝胶状态,动态模量继续随温度升高而快速;
Ⅴ) 150 ℃以上,动态模量随温度升高继续升高,但变化趋势趋缓,并最终趋于恒值.
对比图1a与图1b可以看出,二者反映的基本信息是一致的,但动态模量可以反映出更多的信息,尤其是可以有效评估凝胶化现象.这是因为树脂一旦凝胶化,其流动能力就彻底丧失了,因此,通过该测试可以有效确定凝胶化温度[8-10].
2.2 升温速率对恒温固化的影响
不同升温速率条件下,树脂的黏度对温度的关系如图2左图所示,可以看出,在升温的起始阶段,黏度仅依赖于温度而与升温速率无关,这表明在温度区间,树脂的固化反应几乎可以忽略;但需要注意的是,并不能贸然将黏度开始上升的温度标示为固化反应开始的温度,这是因为该特征温度依赖于升温速率.另外,升温速率越高,最低黏度点对应的温度越高,则最低黏度越低.虽然表面上,似乎提高升温速率可以加大树脂的充模温度窗口,但这并不意味着充模过程中的升温速率越高越好,这是因为升温速率越高,对应的时间窗口越窄,这可以通过对比不同升温速率下升温过程中的黏度对时间的演变进行印证(如图2右图所示).从图2右图可以看出,升温速率越高,黏度降低的窗口越窄,即充模的时间窗口越窄,因此,充模过程中的升温速率选择要综合考虑窗口温度和窗口时间,需要在二者之间做出平衡.
2.3 恒温固化过程中的复数黏度及动态模量演进
在恒温120 ℃条件下,环氧树脂固化过程中体系黏度的改变如图3a所示,可以看出,在初始阶段黏度随时间保持短暂的不变,紧接着快速增加,然后再趋于缓慢增加,最终黏度增加到基本不再改变,总体趋势表现为S型.黏度对时间的演变特征可以分为3个阶段:
Ⅰ) 保温初期(300 s以内),黏度对时间保持短暂不变,表明固化反应存在一定的反应诱导期;
Ⅱ) 保温中期(300~2 100 s),黏度随时间急剧增加,表明固化反应进入快速增长期;
Ⅲ) 保温后期(2 100 s以后),黏度对时间基本不再变化,可能的原因是固化基本完全或者玻璃化高黏度导致了反应动力学禁阻.
在恒温120 ℃条件下,环氧树脂固化过程中体系动态模量的改变如图3b所示.对比图3a与图3b可以看出,动态模量随时间的演变与黏度随时间演变的趋势基本一致,但可以给出比黏度演变更多的信息,主要体现在树脂状态上.对比黏度演变特征,可以将动态模量演变特征分为4个阶段:
Ⅰ) 保温初期(300 s以内),动态模量对时间基本保持不变且G″>G′,表明树脂处于尚未反应的流体态;
Ⅱ) 保温中前期(300~867 s),动态模量对时间保持快速增长,但G″仍然大于G′,表明虽然反应速率较快,但体系仍处于流体态;
Ⅲ) 保温中后期(867~2 100 s),动态模量继续保持快速增长,且G′开始大于G″,表明体系已经凝胶化,丧失了流动能力;

图2 升温速率对固化演进过程的影响
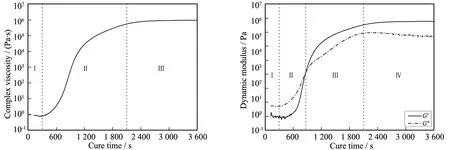
图3 恒温固化过程中的复数黏度(a)和动态模量(b)演变
Ⅳ) 保温后期(2 100 s以后),G′基本保持不变,而G″则呈现缓慢降低.
动态模量随时间演变曲线中,G′=G″对应的时间定义为凝胶时间[7-11],此时交联网络开始形成,体系流动能力丧失,在加工工艺是一个重要的加工参数.
2.4 固化温度对恒温固化过程的影响
不同固化温度(110、115、120、130、140 ℃)条件下恒温固化过程中黏度演变特征如图4a所示,可以看出,随着固化温度的提高,反应诱导期明显缩短,且反应增速期大大提前;但需要注意的是,除110 ℃固化保温外,其他温度下,体系的最终黏度基本趋于一致,表明固化后期的黏度随时间基本不变是由于固化完全所造成而非反应动力学禁阻所致.
不同固化温度(110、115、120、130、140 ℃)条件下恒温固化过程中动态模量演变特征如图4b所示,可以看出,固化温度越高,固化反应诱导期越短,固化反应的速率越快,且达到凝胶点(动态模量交点)和G′基本不变的时间也越短.
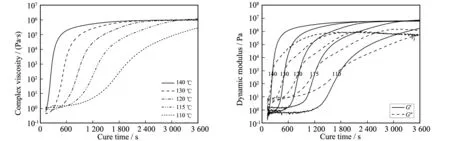
图4 固化温度对恒温固化过程中复数黏度(a)和动态模量(b)演变的影响
3 结论
通过一系列升温测试,获取了树脂复数黏度和动态模量对温度的演变关系,可以初步筛选合适的注射温度区间.通过一系列不同温度的保温测试,获取了树脂复数粘度和动态模量对时间的演变关系,可以进一步筛选加工所需要的合适保温温度及保温时间.本文实验结果表明,黏弹性能可有效示踪树脂固化过程中的结构演变,能为反应成型加工提供有效、有用的工艺参数.
[1] GUO Q.Thermosets [M].Woodhead Publishing Limited, 2012: 3-91.
[2] DEBDATTA R.Handbook of thermoset resins [M].iSmithers, 2009: 155-186.
[3] JOHNANNES K F.Reactive polymers fundamentals and applications [M].William Andrew Inc, 2005: 139-240.
[4] HAN C D.Rheology and processing of polymeric materials Volume 2 [M].Oxford: Oxford Univ Press, 2007: 495-516.
[5] HAN C D.Rheology and processing of polymeric materials Volume 1 [M].Oxford: Oxford Univ Press, 2007: 651-694.
[6] PETER J H.Chemorheology of polymers [M].Cambridge: Cambridge Univ Press, 2009: 321-350.
[7] WINTER H H.Can the gel point of a cross-linking polymer be detected by theG′-G″ crossover [J].Polym Eng Sci, 1987, 27(22): 1698-1702.
[8] PETER J H.Chemorheology of thermosets [J].Polym Eng Sci, 1996, 36(5): 593-609.
[9] MALKEN A Y.Rheokinetics of curing [J].Adv Polym Sci, 1991, 101: 217-257.
[10] HALLEY P J, MACKAY M E, GEORGE G A.Determining the gel point of an epoxy resin by various theological methods [J].High Perform Polym, 1994, 6(4): 405-414.
[11] HATSO I.Handbook of benzoxazine resins [M].Elsevier, 2011: 143-155.
[责任编辑:张普玉]
Rheological monitoring of the reacting curable epoxy resin
LI Runming, TIAN Pei, SHI Jiahua*
(CollegeofChemistryandChemicalEngineering,HenanUniversity,Kaifeng475004,Henan,China)
Epoxy resins form the matrix in filled plastics and fiber-reinforced composites used in a diversity of products.These range from consumer items and auto body panels to advanced composites for printed circuit boards (PCBs), aerospace structural components, and high-performance sports equipment.As the curing reaction progresses, the two moduli cross at the gel point, beyond which the storage modulus becomes larger than the loss modulus and the material hardens.These techniques provide a greater understanding of traditionally complex thermoset processes and effective quality control measures for these processes, and will reduce design and operating costs in associated industries.
epoxy; curing; complex viscosity; dynamic modulus
2016-09-26.
河南省教育厅重点科研项目(14A430013).
李润明(1973-),男,副教授,研究方向为聚合物流变学.*
O631
A
1008-1011(2016)06-0763-04
猜你喜欢
北京航空航天大学学报(2021年9期)2021-11-02
阅读(快乐英语高年级)(2021年11期)2021-03-08
山东交通科技(2020年1期)2020-07-24
山西建筑(2020年11期)2020-06-04
中国盐业(2018年18期)2019-01-14
中国塑料(2016年3期)2016-06-15
浙江大学学报(工学版)(2016年9期)2016-06-05
汽车零部件(2015年1期)2015-12-05
中国塑料(2015年7期)2015-10-14
燕山大学学报(2014年1期)2014-03-11
