
有序介孔碳材料的软模板合成、结构改性与功能化
2016-12-29 08:19胡艳艳屈德宇
物理化学学报 2016年12期
刘 丹 胡艳艳 曾 超 屈德宇
(武汉理工大学化学化工与生命科学学院化学系,武汉430070)
有序介孔碳材料的软模板合成、结构改性与功能化
刘 丹 胡艳艳 曾 超 屈德宇*
(武汉理工大学化学化工与生命科学学院化学系,武汉430070)
有序介孔碳材料在吸附、分离、催化以及能量存储/转化等方面具有广阔的应用前景。相较于复杂的硬模板路线,基于两亲性嵌段共聚物和聚合物前驱体间(如酚醛树脂)自组装的软模板路线是合成有序介孔碳材料更为有效的方法。本文讨论比较了溶剂挥发诱导自组装法、水相协同自组装法和无溶剂法等三种典型软模板路线的基本过程和特点,并介绍了近年来在新型碳前驱体应用、介孔碳的结构改性和功能化等方面的一些重要进展,最后总结了介孔碳的合成研究中所需解决的关键问题。
有序介孔材料;多孔碳;嵌段共聚物;软模板;自组装
1 引言
多孔碳材料具有高比表面积、导电性、化学惰性以及低密度等特点,已在众多科技领域得到广泛应用,如作为吸附剂、色谱分离材料、催化剂载体、储能器件的电极材料等。多孔碳材料(如活性炭和碳气凝胶等)一般通过煤、生物质或者聚合物等前驱体的高温热解并经物理、化学改性制得。然而,通过这些传统的方法一般难以有效控制孔隙结构,获得的多孔碳材料往往包含无序连接、大小不一且形状各异的孔。有序介孔碳材料是近十多年来迅速发展起来的一类多孔碳材料,与传统多孔材料相比,它们具有孔隙排列规则、均匀且易于调控的特点,其一经问世便迅速受到化学、物理及材料学界的高度关注。

刘丹,2002年本科毕业于武汉理工大学化学系,2011年获武汉理工大学材料物理与化学博士学位。现为武汉理工大学化学系副教授。2016年初赴美国威斯康辛大学进行博士后研究。主要从事多孔材料的设计合成及其在电化学储能方面的应用。

胡艳艳,2015年本科毕业于河南工业大学,现在武汉理工大学化学、化工与生命科学学院化学工程与技术专业,科学硕士研究生在读。主要从事介孔碳材料的合成以及电化学性能方面的研究。

曾超,2014年本科毕业于武汉理工大学理学院应用化学专业。同年,进入武汉理工大学化学、化工与生命科学学院应用化学专业进行研究生学习。主要从事有序介孔碳材料合成方法学相关领域研究。

屈德宇,1993年毕业于武汉大学化学系,2002年获加拿大渥太华大学博士学位。而后以NSERC Postdoctoral fellow和JSPS Postdoctoral fellow身份,在加拿大国家研究院(NRC)和日本北海道大学化学系从事研究工作。2007-2011年为韩国高丽大学世宗分校新材料化学系助理教授。现为武汉理工大学化学系教授。主要从事碳材料的合成,锂电池,超级电容器等方面的研究。
有序介孔碳最初采用硬模板法合成,由Ryoo课题组1和Hyeon课题组2提出,其大致过程为:将蔗糖、酚醛树脂、糠醇、乙腈等小分子前驱体通过初始润湿或者气相沉积法填充到硅基有序介孔材料的孔隙结构中,在一定条件下使前驱体转变为高分子聚合物,经高温热解处理后,再通过NaOH或HF腐蚀除去氧化硅而得到介孔碳材料。所得碳材料的有序孔结构通常与母体氧化硅空间群相似,但拓扑结构相反。例如以类蜂窝结构的SBA-15为模板制得的介孔碳CMK-3为纳米短棒阵列结构,其介孔由碳纳米棒间的空隙产生。这类介孔碳材料往往具有较高的比表面积(>1000 m2·g-1)和孔容(>1 cm3·g-1),同时刚性牺牲模板具有支撑作用,经高温处理或催化作用能够获得石墨化或半石墨化的有序介孔碳材料3。相似地,以沸石分子筛为模板可制得有序微孔碳材料(分子筛模板碳)4,5,以胶态晶体为模板可制得有序大孔碳材料6,以无机物为前驱体还可制得各种多孔陶瓷材料7。然而,硬模板法步骤繁琐、成本高昂,且反相复制的孔隙结构稳定性差,极大限制了这些多孔材料的实际应用。
利用软物质,如表面活性剂分子聚集而成的胶团、反胶团、囊泡等作为模板是合成多孔材料和纳米结构材料的另一类重要方法。真正将软模板法成功应用于合成有序介孔高分子及碳材料的是戴胜课题组8、Nishiyama课题组9以及赵东元课题组10的先驱性工作。他们不约而同地选用酚醛树脂为碳源前驱体,两亲性嵌段共聚物为模板剂,通过两者之间的氢键相互作用进行有机-有机自组装获得高分子/嵌段共聚物有序介观复合物,再利用两者之间的热稳性差异,在惰性气氛下热解获得具有开孔结构的有序介孔碳材料。显然,这种基于有机-有机自组装的软模板过程相对于硬模板过程而言大为简化,为介孔碳的大规模工业生产和实际应用提供了可能。
关于介孔碳合成及应用的研究进展国内外已有多位研究者进行了较全面的综述11-19,本文结合本课题组的工作,对介孔碳的软模板合成、结构改性和功能化等方面的研究进展进行了简要介绍。应当指出的是,“介孔碳”的概念实际包含无序孔和有序孔结构的介孔碳,本文中提及的“介孔碳”特指具有有序液晶结构、孔径介于2-50 nm的这类多孔碳材料,无序介孔碳不在本文讨论之内。
2 有序介孔碳的合成方法
相对于硬模板有序介孔碳,以氧化硅为代表的有序介孔无机材料通常采用软模板法获得。其基本原理为通过无机前驱体与表面活性剂分子间的氢键、库仑力或者共价键等相互作用,进行所谓的有机-无机自组装得到液晶相介观复合物,最后除去胶束模板获得对应的产物。这种方法从上世纪九十年代至今已获得长足的发展,形成了蔚为壮观的有序介孔材料大家族。软模板法不需要预制模板材料,显然更为简单易行,该过程中发生的主要化学反应本质上是无机物种间的缩合/聚合反应。因此,人们较早就希望将该过程移植到高分子体系,通过表面活性剂和高分子单体(或者寡聚物)的自组装获得有序介孔高分子材料,再经高温碳化处理后实现介孔碳材料的软模板合成。1999年,Moriguchi等20最早借鉴经典介孔氧化硅MCM-41的合成,以阳离子表面活性剂十六烷基三甲基溴化铵(CTAB)为结构导向剂,尝试与苯酚/甲醛(PF)树脂预聚体进行自组装。由于CTA+阳离子在碱性条件下能够与带负电荷的PF阴离子形成强库仑相互作用,他们成功得到层状和二维六方有序介观结构的胶束/高分子复合物,但热解后仅得到无序非孔性的碳产物。另一些研究者则选用间苯二酚/甲醛(RF)树脂以及硝酸处理的中间相沥青等与CTAB进行自组装,也未成功获得有序介孔结构的碳材料21-23。究其原因,这些“失败”除了未能控制好在介观相形成过程中高分子前驱体自身的聚合反应之外,另一个重要原因是阳离子表面活性剂胶束和高分子骨架间缺乏相溶性,高分子自聚合(未与表面活性剂共组装)导致宏观相分离。另外,离子型表面活性剂分子具有长烷基链,惰性气氛下热解后残碳量高,导致模板除去不完全而造孔失败。
通过有机-有机自组装合成有序介孔高分子和碳材料,模板剂和碳源前驱体的选择最为关键。从模板剂选择的角度来看,在众多类型的表面活性剂中,实际上到目前为止仅两亲性嵌段共聚物适用,且多为含聚氧乙烯(PEO)和聚氧丙烯(PPO)嵌段的Pluronic型嵌段共聚物。除了这类表面活性剂自身的组装特性外,其含氧量较高也至关重要,有利于在惰性气氛高温焙烧过程中发生自氧化而被较完全除去(如Pluronic P123或F127在350-400°C时除去率可达99%(w)24),从而脱除模板获得开孔结构。从碳源前驱体选择角度来看,需至少满足三个条件:(1)前驱体(高分子单体或者寡聚物)能与嵌段共聚物分子通过氢键或者库仑力相互作用;(2)前驱体能在一定条件下聚合形成稳定的刚性高分子网络,且高温热解后具有较高的残碳率,避免模板脱除和碳化过程中发生结构坍塌;(3)聚合反应易于控制,避免前驱体与胶束模板宏观相分离形成非孔结构或者生成低有序度的动力学产物。虽然目前研究者们已尝试了大量可聚合单体作为前驱体,但基于各类酚醛树脂(典型的如PF、RF、间苯三酚/甲醛等)与嵌段共聚物的自组装合成最为成功。酚醛树脂及其单体的化学特性与合成经典有序介孔氧化硅材料所使用的前驱体(典型如正硅酸四乙酯)在许多方面有相似之处,例如在酸或者碱催化下均能通过溶胶-凝胶过程形成三维刚性的网络结构,富含酚羟基的各类酚醛树脂也能像硅酸盐一样与嵌段共聚物通过氢键或者库仑力形成较强的相互作用。同时,固化的酚醛树脂在惰性气氛具有较高的热解残碳率(>50%(w)),能够保证模板脱除和碳化过程中不发生结构坍塌。
2.1 溶剂挥发诱导自组装法
溶剂挥发诱导自组装法(EISA)是以弱极性易挥发的有机溶剂(如乙醇和四氢呋喃等)作为反应介质来制备有序介孔材料的一种重要方法。这种方法一般是将前驱体寡聚物和表面活性剂溶解于有机溶剂中,形成稀溶液,利用溶剂的逐渐挥发诱导微相分离形成复合液晶相,然后经进一步交联固化处理,形成刚性、均匀的介观结构,最后脱除模板得到有序介孔材料。由于该法制备介孔材料经历了典型的溶胶-凝胶过程,最初被用于在基片上沉积制备有序介孔氧化硅薄膜材料25。该法的一个重要特征是前驱体的交联聚合和表面活性剂自组装可认为是分开进行的,技术上避免了前驱体与表面活性剂的协同自组装过程,在必须使用具有较高反应活性前驱体(一般为醇盐)合成非硅基介孔材料的方面表现出独特的优势26,后来逐渐发展为有效制备纯氧化硅、掺杂氧化硅、有机-无机杂化材料、金属氧化物、复合金属氧化物、磷酸盐等各种组分介孔材料的一般方法。
如上所述,与最初合成非硅基介孔材料所遇到的问题一样,由于有机单体在表面活性剂自组装条件下的反应动力学不易控制,利用协同自组装制备有序介孔高分子及碳材料并不成功。2004年,戴胜课题组8率先将EISA法应用于有序介孔碳材料的合成,他们以聚苯乙烯-b-聚乙烯基吡啶(PS-b-P4VP)两嵌段共聚物为结构导向剂,通过PVP嵌段和间苯二酚之间的氢键作用,将间苯二酚和PVP键合,再采用溶剂挥发诱导PS-b-PVP自组装形成有序介观结构,进一步通过熏蒸甲醛以交联间苯二酚得到刚性酚醛树脂骨架,高温碳化并脱除模板PS-b-P4VP后得到具有二维六方结构(p6m)且孔道垂直于基片的有序介孔碳薄膜材料。Nishiyama课题组9则以廉价的、商品化的且已广泛用于介孔无机材料合成的PEO-PPO-PEO型三嵌段共聚物Pluronic F127作为模板剂,以原乙酸三乙酯(EOA)、间苯二酚和甲醛一起作为碳前驱体,并以盐酸作为聚合催化剂,通过典型EISA过程结合旋涂法在硅基片上制备出有序介孔碳薄膜。作者指出,原乙酸三乙酯的引入是提高产物有序性的关键,但并未给出明确解释,可能原因是EOA能减缓高反应活性的间苯二酚与甲醛的聚合速率12。另外,产物虽然具有高度有序的介孔结构,但其对称性解析并不明确。
赵东元课题组10,24进一步将EISA法发展为合成不同结构、不同孔径的有序介孔高分子、碳材料及其掺杂或复合材料的成熟可靠方法。他们以可溶性低聚酚醛树脂(A阶酚醛树脂,resol)作为碳源,与几种常见的Pluronic型三嵌段共聚物(如P123、F127和F108等)在乙醇介质中进行挥发诱导自组装,制备出包括双连续立方(Ia3d,FDU-14)、二维六方(p6mm,FDU-15)、体心立方(Im3m,FDU-16)和层状结构的有序介孔酚醛树脂和碳材料。典型合成过程分为5步(图1):(1)碱(NaOH)催化合成resol;(2)溶剂挥发诱导有机-有机自组装形成resol/嵌段共聚物复合介观相;(3)resol骨架的热固化;(4)低温焙烧脱除模板;(5)高温热解碳化。进一步的研究证明,这种方法具有良好的一般性和普适性,通过选用不同的模板剂和控制条件,可制得多种不同结构、不同孔隙特征且高度有序的介孔碳材料。应当指出的是,该方法中EISA过程是在近中性条件下完成,自组装驱动力源于三嵌段共聚物中PEO嵌段与酚醛树脂中酚羟基之间的氢键相互作用。戴胜课题组27还在强酸性条件下(0.5-2.0 mol·L-1HCl),以RF为前驱体,F127为模板剂,采用乙醇/水(1/1,V/V)混合物作为反应介质,成功制备了有序介孔碳(C-ORNL-1)。与合成介孔碳FDU-14等先单独制备PF酚醛树脂不同,合成C-ORNL-1时则是先制备RF/F127复合物,通过离心分离后再溶解于四氢呋喃中进行EISA过程。由于强酸性条件,作者认为RF树脂和F127均被质子化,因此组装驱动力为两者间的库仑相互作用。该法相对更为简单,而且获得介孔碳材料具有高热稳定性,但仅能制得简单二维六方结构的介孔碳。

图1 采用EISA法制备不同结构的有序介孔树脂和碳材料24Fig.1 Schematic illustration for preparation of ordered mesoporous resins and carbons via solvent evaporation-induced self-assembly(EISA)24
一般认为,EISA合成有序介孔材料的过程中,前驱体聚合和表面活性剂自组装这两个过程是分开进行的28。正如上述介孔碳FUD-x的合成过程中,在溶剂乙醇挥发之前,嵌段共聚物的亲水端(PEO嵌段)和疏水端(PPO嵌段)都能溶解于乙醇中,并不表现出两亲特性,因此不会发生自组装形成聚集体(胶束),嵌段共聚物主要以个体分子分散于溶剂中。随着溶剂的挥发,体系中嵌段共聚物和PF低聚物浓度逐渐增大,亲水PEO嵌段通过氢键作用逐渐溶解于PF树脂网络中,自组装发生,最后达到亲疏水平衡形成复合液晶结构,之后通过加热处理使PF树脂进一步交联固化形成稳定的三维网络结构。由此可见,苯酚与甲醛缩聚形成高分子酚醛树脂分两步完成,一是溶剂挥发之前的预聚(形成分子量小于500的可溶性低聚酚醛树脂),二是溶剂挥发之后的加热固化(形成高分子酚醛树脂)。另外,由于中性介质且在室温进行,在溶剂挥发过程酚醛树脂难以发生交联反应。所以,EISA法的最大优点是技术上避免了协同自组装过程,使合成中涉及的两个过程分阶段进行,从而有利于对各个过程进行独立控制。也正因如此,软模板介孔碳的成功合成绝大多数基于EISA法。必须提及的是,Schuster等29利用小角X射线散射(SAXS)技术对合成介孔碳FDU-x进行了原位研究,发现仅在加热固化一段时间后才能观测到对应介观结构的SAXS信号,因此认为有序液晶结构的形成发生在加热固化阶段,而不是溶剂挥发阶段。该发现颠覆了对EISA过程的原有认知,假如确实如此,该方法应改名为热诱导自组装法(TISA)。然而,笔者认为,在未加热之前观测不到信号也有可能是此时低聚酚醛树脂结构还比较“松散”,与同为有机物的嵌段共聚物胶束间的电子密度衬度较低造成的。总之,该观点仍需采用其它技术进行进一步佐证。
虽然采用EISA法在合成不同结构和不同性质的有序介孔碳及其相关材料方面取得了巨大成功,相对于传统硬模板法具有极大的优势,EISA法也有其固有的缺点,比如需要大面积铺膜以及消耗大量有机溶剂且难以回收,实际上也面临着难以大规模工业生产的难题。
2.2 水相协同自组装法
经典有序介孔氧化硅材料(如MCM-41、SBA-15和KIT-6等)的合成多采用水相协同自组装法。相对于非水EISA法,这种合成方法至少具有以下几个优点:(1)在稀水溶液中,有序液晶相的形成可主要受热力学控制,能够得到缺陷少且高度有序的产物;(2)水相合成本质上是共沉淀过程,不需大面积铺膜工艺,合成不受批尺寸的限制,适用于大批量工业化生产;(3)产物形貌丰富且可控;(4)以水为反应介质,更为绿色、经济。
由于包含复杂的协同自组装过程,相对于硬模板法和EISA法,水相合成有序介孔碳的成功例子相对较少。赵东元课题组30,31在稀水溶液中,以NaOH为催化剂,利用低分子量PF树脂预聚物和三嵌段共聚物(F127和P123)自组装也制备出具有双连续立方(Ia3d)、二维六方(p6mm)或体心立方(Im3m)的介孔碳。这种水相自组装的驱动力同样源于酚醛树脂和嵌段共聚物间的氢键作用,因此对pH要求较高,只有将体系pH值控制在8.5-9.0时,才能获得有序产物。当pH值过高时,酚醛树脂将转为对应的阴离子,无法与嵌段共聚物形成有效的氢键相互作用;当pH值过低时,酚醛聚合速率太慢。同时,由于嵌段共聚物的浊点限制,反应温度控制在65-70°C。应该指出的是,该合成没有直接使用分子单体(苯酚和甲醛)作为起始反应物,而是使用反应活性较低的PF寡聚物与嵌段共聚物进行自组装,这种策略有效避免了分子单体间不可控的缩合/聚合反应,可使反应在近热力学控制的条件下进行,甚至能够制备出有序度极高的“单晶”介孔碳32,33。该合成开辟了在稀水溶液中合成不同结构且高度有序的介孔高分子和碳材料的先例,为其大规模生产和应用奠定了基础。然而,其缺点是反应周期较长,整个溶液反应需在65-70°C进行5天以上。陆安慧等34以谷氨酸作为催化剂,以间苯二酚和甲醛单体作为起始物,与F127进行水相自组装制备出较低有序性的二维六方介孔碳。谷氨酸不仅作为酚醛缩合的弱酸催化剂,还参与酚醛聚合形成聚苯并噁嗪骨架。该方法直接使用分子单体,但需额外的热固化处理,合成周期也较长(包括溶液反应和热固化需6天)。
以上的合成均是在常压条件下进行的,研究者们也探索了在高压反应釜中水热合成有序介孔碳。在反应物浓度较高的情况下,可制备单块状产物35,36;在反应物浓度极低的情况下,则可制备有序介孔碳纳米粒子37,38。由于水热处理温度较高(≥100°C),只能选择高浊点的嵌段共聚物(如F127)作为模板,且反应受动力学控制,产物有序度相对较低,但可大大缩短反应时间。
基于前人的工作,我们提出了基于酚/六次甲基四胺为前驱体对在水相合成有序介孔碳的新方法39。该法十分简单,仅需将间苯二酚、六次甲基四胺(HMT)、模板剂(F127)以及氨水在室温下搅拌溶解于水,然后在80°C加热反应24 h完成溶液反应,碳化后即可得到高质量的体心立方结构的有序介孔碳材料。间苯二酚是水溶性最好的酚类化合物之一,其与甲醛的反应活性大约是苯酚的10-15倍40。通过有机溶胶-凝胶过程得到的RF气凝胶已广泛用于碳气凝胶的制备41。然而,间苯二酚与甲醛的聚合反应十分复杂,对R/F比、催化剂浓度、pH值、温度甚至加料顺序等条件变化均极为敏感。事实上,采用表面活性剂自组装法合成介孔碳最初就是以RF作为前驱体,但是仅仅获得了无序材料21,23,重要原因之一是未能很好的控制高反应活性的间苯二酚与甲醛的聚合反应12。我们未从调节反应温度、催化剂用量以及反应物浓度等常规手段来解决RF聚合反应不易控制的难题,而是选用HMT代替甲醛作为交联剂。HMT在非酸性、近室温条件的水溶液中十分稳定,但当温度高于70°C时,则会缓慢水解释放出甲醛和氨(见图2)。实际上,HMT在纳米材料的控制合成方面最著名的例子是作为碱(氨)缓释剂制备氧化锌纳米线阵列42。在我们的合成体系中,HMT则作为甲醛的缓释剂。在优化的反应温度(80°C)条件下,HMT水解反应能够作为决速步骤(RDS)使整个协同自组装过程在近热力学控制的条件下进行,从而获得了高度有序的产物。当我们试图在反应体系中加入一种常用的介孔扩孔剂间三甲苯(TMB)时,发现产物结构发生了相变,从而还成功获得了二维六方结构的介孔碳。与以前的方法相比,我们的方法具有简单易行、产物结构规整、不引入无机杂质、适于批量制备等优点。在后续的研究中,我们还发现该介孔碳中还含有少量氮元素(~1%(x))43,可能原因是氨参与了酚醛聚合反应而形成聚苯并噁嗪,碳化后产物实际上为氮掺杂的有序介孔碳。

图2 以间苯二酚/六次甲基四胺为前驱体对合成有序介孔碳39Fig.2 Schematic illustration for synthesis of ordered mesoporous carbons by using resorcinol/hexamine as a precursor pair39
我们的方法在合成不同结构、形貌以及功能化的介孔碳方面也具有良好的普适性。例如,将反应在高压反应釜中进行,可制得有序介孔碳球44。最近,我们将该体系中催化剂氨替换为碱性氨基酸(L-赖氨酸或者L-精氨酸),发现能够将最优反应温度降低至70°C;同时,在保持产物高度有序的前提下,可对产物的氮含量、形貌和结构进行调控45。另一些研究者则将间苯二酚简单替换成其它酚来制备功能性介孔碳材料,如换用成3-氨基酚,则可获得具有高含量氮掺杂的介孔碳纳米球46-48和单晶49;如部分换成4-氟苯酚,则可获得氟掺杂的介孔碳50。Lu等51最近发现在间苯二酚与HMT聚合反应之前,引入间苯三酚和对苯二甲醛,经水热聚合-碳化合成出含氮超微孔碳纳米球。
前已提及,酚醛树脂与硅酸盐(如正硅酸乙酯)有许多相似的化学性质。例如,它们的聚合反应既能被酸催化,也能被碱催化。近期报道的有序介孔碳的水相合成多是在弱碱性条件下进行,自组装驱动力是酚醛树脂和嵌段共聚物间的单层氢键相互作用12。相对地,有序介孔氧化硅基材料(如SBA-15、SBA-16和KIT-6等)则多在强酸性条件下,通过质子化的硅酸盐和质子化的共聚物间的库仑相互作用进行自组装得到28。这启发我们尝试在强酸性水溶液中,以间苯二酚/六次甲基四胺为前驱体对合成有序介孔碳,并获得了成功52。有趣的是,该合成与经典有序介孔氧化硅SBA-15的合成有诸多相似之处,主要表现在以下3个方面:(1)使用浓度相近(2.6%-4.0%(w))的P123为模板剂,获得的产物均为二维六方对称(p6m)结构且具有相似的纤维状形貌(见图3);(2)最优盐酸浓度为~2 mol·L-1,与典型SBA-15合成相近(~1.6 mol·L-1);(3)组装驱动力为库仑力。另外,通过改变反应条件,如P123浓度和反应温度,可调控碳材料的尺寸及其它结构性质。强酸性合成路线相对于碱性合成路线也具有一些优点:(1)反应温度相对较低(酸性条件降低了HMT的水解温度),使具有较低浊点的模板剂(如P123)也得以适用,从而为获得新结构的介孔碳奠定了基础;(2)酚醛树脂在酸性条件下易形成柔性线状结构,有利于获得形貌复杂多变的产物;(3)适于在体系中加入金属离子,为直接合成金属、氧化物或者碳化物掺杂的介孔碳材料提供了可能性。

图3 在强酸性水溶液中合成的纤维状有序介孔碳52Fig.3 Fiberlike ordered mesoporous carbons synthesized in strongly acidic aqueous media52
2.3 无溶剂法
无溶剂法是合成另外两类重要多孔材料——微孔沸石分子筛53和金属有机骨架材料54的重要方法。最近,Wang等55首次将无溶剂法应用到有序介孔碳材料合成中。其主要过程为:首先采用机械研磨的方法将间苯二酚、对苯二甲醛和嵌段共聚物在固相条件下混匀,再经过100°C热处理,使混合物熔融并交联固化,最后在600°C焙烧以除去表面活性剂和碳化,从而得到有序介孔碳(图4)。作者指出该法其实并不是绝对的无溶剂条件,因为间苯二酚具有较强吸湿能力,在研磨过程中会吸附~50%(w)水分。虽然如此少量的水不足以溶解表面活性剂和前驱体,但是能有效的促进酚醛前驱体与F127间的氢键作用力,有利于有序介观结构的形成。该法“三废”排放少,原料几乎完全参与反应,符合绿色化学和原子经济性的要求。该法另一优点是可方便实现介孔碳的掺杂,如研磨时加入三聚氰胺,可得氮含量高达24.4% (w)的氮掺杂介孔碳材料55-57;加入磷钼酸可获得碳化钼掺杂介孔碳材料55;加入三聚氰胺和硝酸镁可获得氮、氧化镁共掺杂的介孔碳材料58。无溶剂法虽然具有上述众多优点,但产物有序度不是太高,且热稳定性较差,在高于临界碳化温度(600°C)焙烧后产物结构会发生严重坍塌。另外,其具体的机理尚不明确。比如,有序介观相形成于何阶段?是如Schuster等29所认为的热诱导形成吗?这些问题还有待解答。

图4 无溶剂法合成有序介孔碳55Fig.4 Schematic illustration for solvent-free synthesis of ordered mesoporous carbons55
3 非酚醛树脂基有序介孔碳
各类酚醛树脂被证明是软模板合成有序介孔碳最为有效的前驱体,然而酚类化合物和甲醛均具有毒性,甲醛更是公认的强致癌物,因此,寻找一些环境较友好的替代前驱体也引起了研究者们的兴趣。
生物质(典型如碳水化合物)的水热碳化(HTC)是合成多孔碳材料一种重要方法59,60。这些生物质一般富含羟基,能与Pluronic型嵌段共聚物发生氢键相互作用,可能是合成介孔碳材料的一类理想的酚醛树脂替代物。但HTC温度多超过150°C,此时多数嵌段共聚物不能稳定存在或者超过了其浊点。2011年,Kubo等61发现D-果糖具有较低的HTC温度,将其与F127溶解于水并在130°C水热处理3-5天后,成功获得了体心立方介观结构的复合物,其晶胞参数达23.6 nm。进一步在氮气气氛下、550°C焙烧脱除模板后,产物晶胞参数收缩至17.4 nm,且保持了高度有序的结构,但其孔径仅为0.9 nm,产物实际为有序微孔碳材料。在体系中加入扩孔剂(1,2,4-三甲苯)后,其孔径可增大至4.0 nm。该介孔碳材料含有丰富的含氧基团(如酚羟基和羰基等),但其比表面积和孔容分别仅为116 m2·g-1和0.10 cm3·g-1,均远逊于酚醛树脂基介孔碳。Feng等62以β-环糊精为碳源,以F127和P123为混合模板,在140°C水热处理并在700°C热解处理后,得到了高度有序的二维六方介孔碳,其比表面积和孔容分别可达781 m2·g-1和0.41 cm3·g-1,与FDU-15相当。值得提及的是,该合成体系中额外加入了少量盐酸(~0.2 mol·L-1),但作者并未阐明其作用。最近,Xu等63通过详细研究几种糖前驱体(木糖、阿拉伯糖和核糖等)和F127的自组装体系,发现酸能催化HTC过程,从而降低糖类物质的HTC温度同时提高HTC反应速率。
除碳水化合物外,Schlienger等64选择单宁酸为前驱体,F127为模板,通过EISA法制备出有序度较低的介孔碳。由于单宁酸在酸性近室温条件下可发生自聚合,因此无需加入交联剂甲醛。最近,Braghiroli等65进一步实现了在水溶液中利用单宁酸与F127的自组装合成介孔碳。溶液反应可在室温条件下进行,同时在0至4.2的较宽的pH值条件下均能获得有序产物(见图5)。
除上述生物质外,鲍哲楠课题组66最近还选用一种吡咯衍生物作为前驱体,P123为模板剂,FeCl3为催化剂,在酸性水溶液中制备出有序介观结构的聚吡咯,碳化后获得氮掺杂的有序介孔碳材料。由于聚吡咯是最受关注的导电高分子之一,该方法的重要意义在于首次采用软模板法实现了有序介孔导电高分子材料及其相应碳材料的制备。
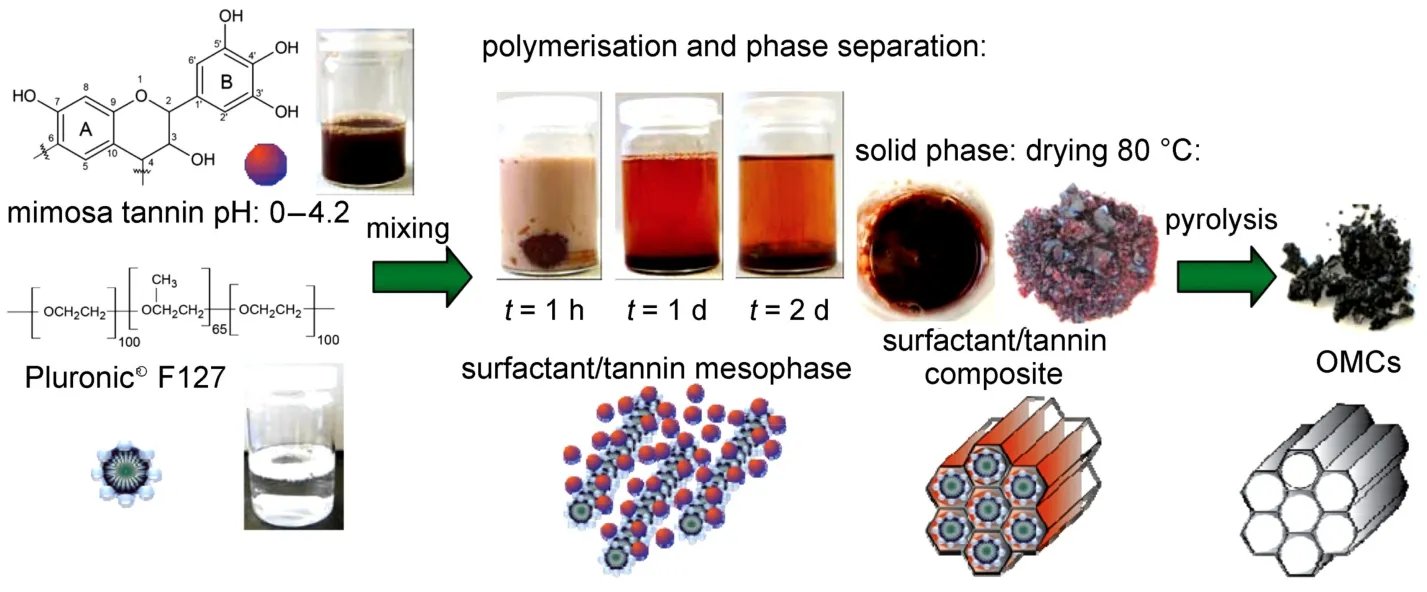
图5 在水溶液中利用单宁酸和F127自组装合成有序介孔碳65Fig.5 Schematic illustration for aqueous synthesis of mesoporous carbons by self-assembly of tannin and F12765
4 有序介孔碳的结构改性
相对于硬模板介孔碳,软模板介孔碳通常具有相对较低的比表面积(<1000 m2·g-1)和孔容(<1 cm3·g-1),这极大限制了其在吸附、催化、储能等诸多方面的应用,因此,在介孔结构基础上通过一定手段引入次级孔(包括微孔、介孔和大孔等),从而获得多级孔碳材料成为近年来的研究热点。一般而言,引入微孔更有利于提高材料的比表面积,而引入次级介孔和大孔更有利于提高材料的孔容。
在介孔碳骨架中引入微孔或者小介孔常采用各种活化法,如用KOH67,68、H2O69、NH370,71以及NaNH272等在高温下与介孔碳作用,利用氧化还原反应腐蚀掉一部分碳基体,从而获得微/介多级孔碳材料。其比表面积可高于3000 m2·g-1,孔容可高于2 cm3·g-1,一些方法还能在碳骨架中引入N、O等杂原子。然而,这些活化法的缺点是易造成介孔有序性的严重破坏,同时会不可避免地引入大量不规则孔,如锲型孔、瓶状孔甚至封闭孔等,可能会影响其在一些方面的应用性能,如作为电极材料时,电解液难以完全进入这些孔,使其实际具有较低的电活性表面积。
在制备介/大多级孔碳材料方面,一般可结合应用软、硬模板法实现。产生大孔的模板材料可以是胶态晶体73,74、泡沫75、多孔阳极氧化铝膜76、生物模板77等。图6以胶态晶体作为大孔模板为例,结合采用软模板法制备有序介/大孔材料。基本过程是将嵌段共聚物和碳源前驱体的混合物溶胶渗透至大孔模板的孔隙中,再通过EISA和固化过程在大孔模板孔隙中形成复合液晶相,最后碳化并除去软、硬两种模板得到产物。值得指出的是,刚性的大孔模板通常会抑制液晶相在高温碳化过程中的结构收缩,导致多级孔碳产物中介孔孔径相对普通介孔碳的孔径大一些。同时,这种空间限制效应有时还会引起介孔排列方向的扭曲。
除上述方法外,另一种制备有序多级孔碳材料是所谓的氧化硅辅助合成法79,80,其基本原理是在嵌段共聚物与酚醛树脂的组装体系中,加入预水解的硅酸盐,通过三元共组装制备有序介孔氧化硅/碳复合物,蚀除氧化硅组分后得到了含均匀主孔道和孔壁次级孔的多级孔碳材料(图7)。该类材料不仅可保持高度有序的介孔结构,同时具有较高的比表面积(可达2400 m2·g-1)及孔容(可达2.3 cm3·g-1),但由于孔壁次级孔源于多分散的氧化硅胶体,其尺寸大小不一。另外,Yu81和Liu82等利用低聚酚醛树脂、柠檬酸钛和F127进行三元自组装制备有序介孔碳化钛/碳复合材料,再经高温氯化后获得含介孔结构的微孔碳化物衍生碳(CDCs)材料。这种多级孔碳材料具有CDCs类材料特征的均匀微孔结构,但碳化物结晶和氯气高温处理易造成有序介孔结构的坍塌,同时需在高温条件下使用高毒性、高腐蚀性氯气。

图6 结合软、硬模板法合成有序介/大孔材料78Fig.6 Schematic illustration for synthesis of ordered mesoporous/macroporous materials by a combined soft-and hard-template method78

图7 三元共组装合成有序多级孔碳和氧化硅材料79Fig.7 Schematic illustration for triconstituent co-assembly to hierarchically ordered mesoporous carbon and silica materials79
最近,我们提出了一种基于多面体低聚倍半硅氧烷(POSS)与嵌段共聚物自组装合成有序微/介多级孔碳材料的新方法(图8)83。POSS化合物是一类化学通式为(RSiO1.5)n的有机硅烷化合物(其中R代表有机官能团,n为偶数,典型n=8),具有分子内有机/无机杂化的结构特点,即无机硅氧核周围连接有有机取代基R。POSS常被看作是“最小的氧化硅粒子”,由于其特殊的组成和结构,目前被作为一类新型纳米填料而广受研究者关注84。为了与Pluronic型嵌段共聚物形成氢键作用,我们选择了一种胺苯基功能化的POSS,即笼型八(胺苯基)倍半硅氧烷(OAPS),通过常规的EISA过程获得有序介观结构,在惰性气氛中焙烧之后,嵌段共聚物被脱除,而相互交联的OAPS将转化成氧化硅相和碳骨架。在腐蚀除去氧化硅相后,所得的碳材料不仅拥有POSS“印迹”的均匀微孔(~1 nm),同时还具有高度有序的介孔(~4 nm),且其比表面积和孔容分别可达2100 m2·g-1和1.2 cm3·g-1。通过采用具有不同PEO/PPO比的嵌段共聚物,材料的介观对称性可以为二维六方(p6m)和体心立方(Imm)。另外,因为OAPS前驱体本身富氮,所得的多级孔碳材料还具有较高的掺杂氮含量(~4%(w))。
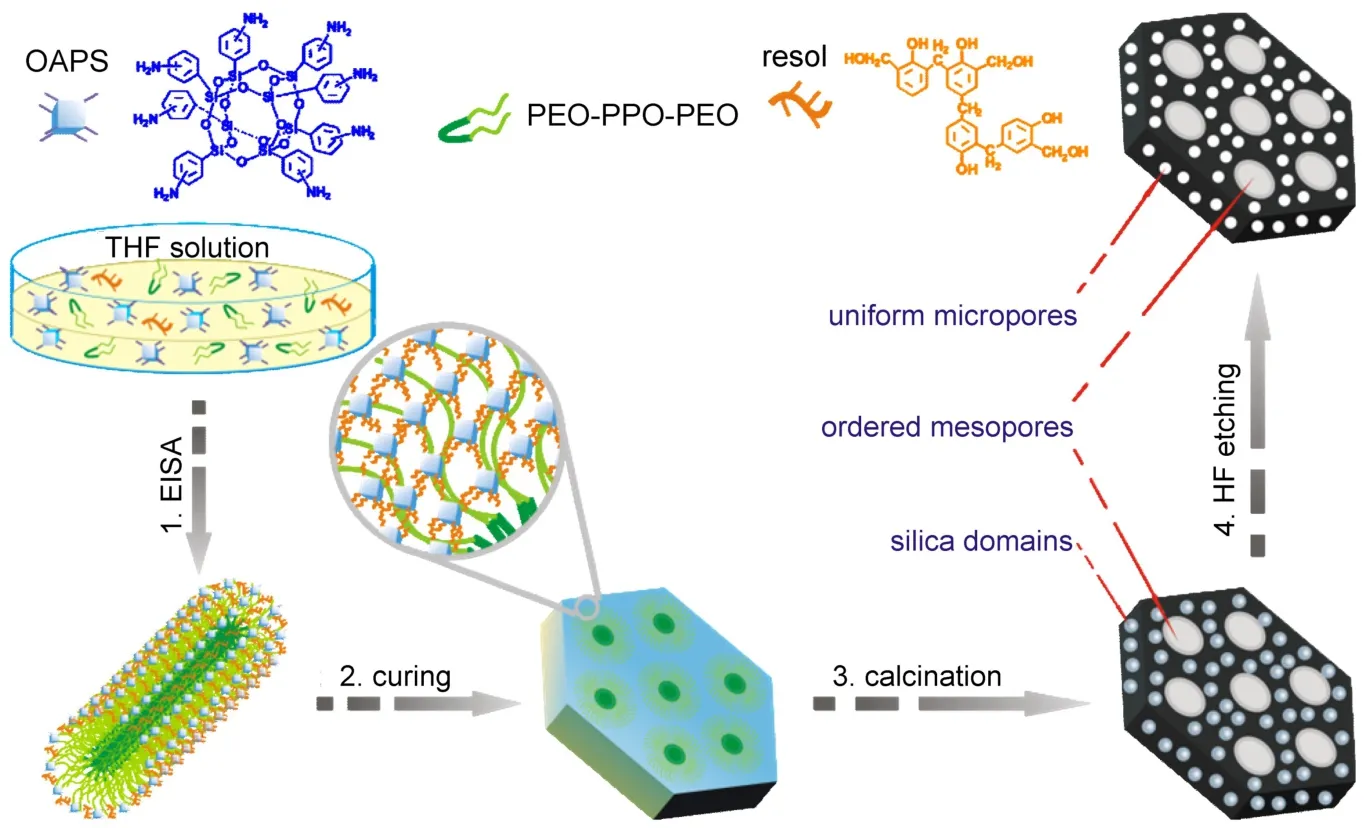
图8 POSS与嵌段共聚物自组装合成有序微/介多级孔碳材料83Fig.8 Schematic illustration for synthesis of hierarchically ordered micro-/mesoporous carbons by self-assembly of POSS and block copolymers83
5 有序介孔碳的功能掺杂
为了扩大应用范围,表面改性和异质原子功能化是多孔碳材料研究领域的一个重要内容,包括非金属元素(如B、N、O、P、S等)和金属元素(如单质、氧化物和碳化物等)均能通过一定方法引入到多孔碳材料的晶格、骨架或者孔隙中。其中,作为在元素周期表中与碳相邻元素之一,碳材料的氮掺杂不仅能够最小化晶格失配,同时也可以改变材料的电子结构,从而带来新的功能特性。例如,氮进入碳晶格能够引入催化活性位点,氮掺杂的多孔碳可作为非(贵)金属催化剂用于有机合成、电催化氧还原(ORR)以及电催化氢析(HER)等85,86;当多孔碳作为超级电容器电极材料时,氮掺杂能够提供除双电层电容外额外的赝电容,同时改善其导电性,从而大幅提高多孔碳电极材料的能量密度和功率密度87,88;当氮掺杂多孔碳用于锂硫电池的载硫阴极材料时,可与极性可溶多硫化物发生强化学吸附作用,从而抑制穿梭效应,改善电池的循环稳定性89,90;此外,氮掺杂还可改善多孔碳在储氢、二氧化碳吸附等方面的性能91。一般而言,氮原子掺杂到碳材料后,在碳原子的晶格中通常会形成四种键合类型(图9):吡咯氮(N-5)、吡啶氮(N-6)、石墨氮(N-Q)以及吡啶氧化氮(N-X)92。热解温度和氮源通常会影响这四类键合氮在碳材料中含量和比例。
有序介孔碳的氮掺杂一般可通过两种方法实现。一是用氨气70,71和三聚氰胺93等富氮前驱体处理预形成的介孔碳或高分子材料;二是使用富氮有机物作为前驱体,通过自组装原位引入。后者显然更为简单,本文主要对这种方法进行简要介绍。Wei等94以Pluronic F127为模板,A阶酚醛树脂为碳源,双氰胺为氮源,采用EISA法合成出氮掺杂有序介孔碳。增加双氰胺的加入量,可将产物中氮含量提高至13.1%(w)。但同时,由于双氰胺和嵌段共聚物间缺乏有效相互作用,会干扰有序自组装,获得的氮掺杂介孔碳有序度较低,且比表面积仅为~500 m2·g-1。经KOH活化后,可将材料的比表面积提高至1417 m2·g-1同时保持相对较高的氮含量(6.7%(w)),但有序孔结构遭到进一步破坏。Song等95进一步在以上合成体系中加入硅溶胶,通过多元自组装制备出氮掺杂有序微/介多级孔碳材料,其比表面积和氮含量分别可达1374 m2·g-1和5.74%(x)。以上方法均基于非水相EISA法,更优的方法是基于水相自组装法和无溶剂法,这方面的研究上文已有提及,不再赘述。
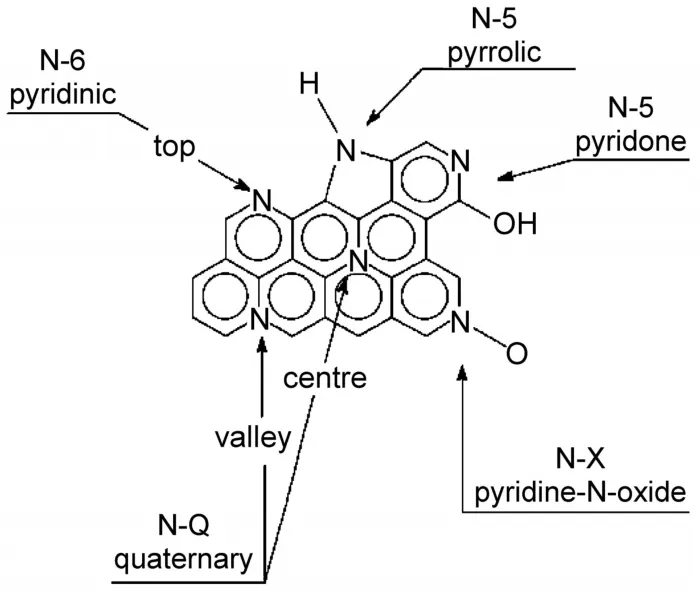
图9 氮在碳晶格中的键合类型92Fig.9 Bonding configurations for nitrogen element in carbon lattice92
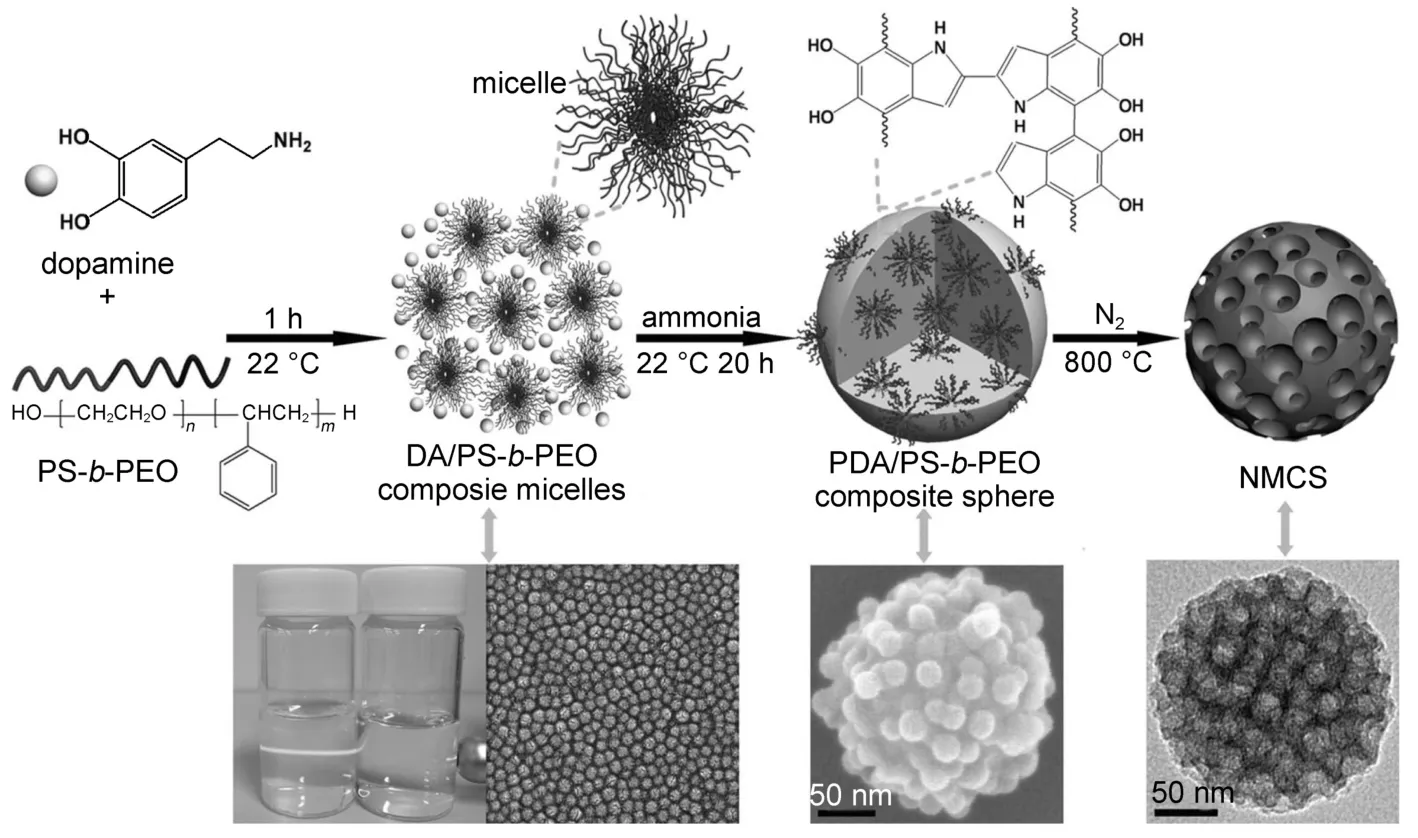
图10 氮掺杂介孔碳纳米球的形成96Fig.10 Formation of nitrogen-doped mesoporous carbon nanospheres96
最近,Tang等96以多巴胺作为碳源和氮源,聚苯乙烯-b-聚氧乙烯两嵌段大分子共聚物为模板剂,在乙醇/四氢呋喃/水混合溶剂中,通过协同自组装制备出氮掺杂有序介孔碳纳米球(图10)。该碳纳米球不仅具有良好的尺寸单分散性(~200 nm)和较大的介孔(~16 nm),其氮含量也可达~7.5%(w)。如果在该合成体系中加入氧化硅纳米球(尺寸约~350 nm),还可制备出含大介孔的氮掺杂介孔空碳球97。
除氮掺杂介孔碳外,另一些非金属异质原子也可通过软模板法掺杂到介孔碳中。例如,Zhu等98将羟基乙叉二膦酸加入到F127/RF组装体系中,制备出磷掺杂的有序介孔碳材料,其比表面积达815 m2·g-1,磷含量为1.62%(x)。Deng等99则以三苯基膦作为磷源,并经氨气活化处理,制备出磷和氮共掺杂的介孔碳,其氮含量为3.32% (x),但磷含量仅为0.16%(x)。Zhao等100在F127/ RF组装体系中加入硼酸和磷酸,合成出硼和磷共掺杂的介孔碳,硼和氮的掺量分别可达1.6%和3.6%(w)。
6 结论和展望
经过十多年迅猛发展,有序介孔碳材料的合成技术日臻完善,人们对其形成机理也有了较为完整、深刻的认识。结合文献以及当前的研究进展,今后在有序介孔碳合成研究方面应关注以下几点:(1)虽然有序介孔碳的结构控制、形貌控制以及功能化等方面已取得了巨大进展,但多采用硬模板法和EISA法实现。水相合成和无溶剂合成是适合工业化大批量制备的方法,但上述方面的研究明显不足。(2)仅利用普通表面活性剂和扩孔剂(如间三甲苯),介孔氧化硅的孔径可从2 nm调节至60 nm以上101。但目前要制备孔径大于10 nm的软模板介孔碳,必须使用非商品化的大分子嵌段共聚物(分子量数万)15。如何利用常见的扩孔剂和商品化的嵌段共聚物作为共模板,调控介孔碳的孔径进而制备大介孔碳材料是值得关注的问题。(3)发展利用生物质为碳源前驱体合成有序介孔碳的方法。(4)在许多应用如电化学储能、催化、吸附等方面,碳材料必须同时具有高掺杂量和高孔性,但目前的合成方法还无法兼顾,亦即高掺杂的介孔碳一般具有较低的比表面积(或孔容),或者相反。
(1) Ryoo,R.;Joo,S.H.;Jun,S.J.Phys.Chem.B 1999,103,7743. doi:10.1021/jp991673a
(2) Lee,J.;Yoon,S.;Hyeon,T.;Oh,S.M.;Kim,K.B.Chem. Commun.1999,2177.doi:10.1039/a906872d
(3) Kim,T.W.;Park,I.S.;Ryoo,R.Angew.Chem.Int.Edit.2003, 42,4375.doi:10.1002/anie.200352224
(4) Ma,Z.X.;Kyotani,T.;Tomita,A.Chem.Commun.2000, 2365.doi:10.1039/b006295m
(5) Kim,K.;Lee,T.;Kwon,Y.;Seo,Y.;Song,J.;Park,J.K.;Lee, H.;Park,J.Y.;Ihee,H.;Cho,S.J.;Ryoo,R.Nature 2016,535, 131.doi:10.1038/nature18284
(6) Yu,J.S.;Kang,S.;Yoon,S.B.;Chai,G.J.Am.Chem.Soc. 2002,124,9382.doi:10.1021/ja0203972
(7) Yang,H.F.;Zhao,D.Y.J.Mater.Chem.2005,15,1217. doi:10.1039/b414402c
(8) Liang,C.;Hong,K.;Guiochon,G.A.;Mays,J.W.;Dai,S. Angew.Chem.Int.Edit.2004,43,5785.doi:10.1002/ anie.200461051
(9) Tanaka,S.;Nishiyama,N.;Egashira,Y.;Ueyama,K.Chem. Commun.2005,2125.doi:10.1039/b501259g
(10) Meng,Y.;Gu,D.;Zhang,F.;Shi,Y.;Yang,H.;Li,Z.;Yu,C.; Tu,B.;Zhao,D.Angew.Chem.Int.Edit.2005,44,7053. doi:10.1002/anie.200501561
(11) Liang,C.;Li,Z.;Dai,S.Angew.Chem.Int.Edit.2008,47, 3696.doi:10.1002/anie.200702046
(12) Wan,Y.;Shi,Y.;Zhao,D.Chem.Mater.2008,20,932. doi:10.1021/cm7024125
(13) Zhai,Y.;Dou,Y.;Zhao,D.;Fulvio,P.F.;Mayes,R.T.;Dai,S. Adv.Mater.2011,23,4828.doi:10.1002/adma.201100984
(14) Chuenchom,L.;Kraehnert,R.;Smarsly,B.M.Soft Matter 2012,8,10801.doi:10.1039/c2sm07448f
(15) Deng,Y.;Wei,J.;Sun,Z.;Zhao,D.Chem.Soc.Rev.2013,42, 4054.doi:10.1039/c2cs35426h
(16) Fang,B.;Kim,J.H.;Kim,M.S.;Yu,J.S.Acc.Chem.Res. 2013,46,1397.doi:10.1021/ar300253f
(17) Ma,T.Y.;Liu,L.;Yuan,Z.Y.Chem.Soc.Rev.2013,42,3977. doi:10.1039/c2cs35301f
(18) Liu,J.;Wickramaratne,N.P.;Qiao,S.Z.;Jaroniec,M.Nat. Mater.2015,14,763.doi:10.1038/nmat4317
(19) Xin,W.;Song,Y.H.RSC Adv.2015,5,83239.doi:10.1039/ c5ra16864c
(20) Moriguchi,I.;Ozono,A.;Mikuriya,K.;Teraoka,Y.;Kagawa, S.;Kodama,M.Chem.Lett.1999,1171.doi:10.1246/ cl.1999.1171
(21) Lee,K.T.;Oh,S.M.Chem.Commun.2002,2722. doi:10.1039/b208052d
(22) Li,Z.J.;Yan,W.F.;Dai,S.Carbon 2004,42,767. doi:10.1016/j.carbon.2004.01.044
(23) Nishiyama,N.;Zheng,T.;Yamane,Y.;Egashira,Y.;Ueyama, K.Carbon 2005,43,269.doi:10.1016/j.carbon.2004.09.009
(24) Meng,Y.;Gu,D.;Zhang,F.;Shi,Y.;Cheng,L.;Feng,D.;Wu, Z.;Chen,Z.;Wan,Y.;Stein,A.;Zhao,D.Chem.Mater.2006, 18,4447.doi:10.1021/cm060921u
(25) Lu,Y.F.;Ganguli,R.;Drewien,C.A.;Anderson,M.T.; Brinker,C.J.;Gong,W.L.;Guo,Y.X.;Soyez,H.;Dunn,B.; Huang,M.H.;Zink,J.I.Nature 1997,389,364.doi:10.1038/ 38699
(26) Yang,P.D.;Zhao,D.Y.;Margolese,D.I.;Chmelka,B.F.; Stucky,G.D.Nature 1998,396,152.doi:10.1038/24132
(27) Wang,X.;Liang,C.;Dai,S.Langmuir 2008,24,7500. doi:10.1021/la800529v
(28) Wan,Y.;Zhao,D.Chem.Rev.2007,107,2821.doi:10.1021/ cr068020s
(29) Schuster,J.;Kohn,R.;Doblinger,M.;Keilbach,A.; Amenitsch,H.;Bein,T.J.Am.Chem.Soc.2012,134,11136. doi:10.1021/ja208941s
(30) Zhang,F.;Meng,Y.;Gu,D.;Yan,Y.;Yu,C.;Tu,B.;Zhao,D. J.Am.Chem.Soc.2005,127,13508.doi:10.1021/ja0545721
(31) Zhang,F.Q.;Meng,Y.;Gu,D.;Yan,Y.;Chen,Z.X.;Tu,B.; Zhao,D.Y.Chem.Mater.2006,18,5279.doi:10.1021/ cm061400+
(32) Zhang,F.;Gu,D.;Yu,T.;Zhang,F.;Xie,S.;Zhang,L.;Deng, Y.;Wan,Y.;Tu,B.;Zhao,D.J.Am.Chem.Soc.2007,129, 7746.doi:10.1021/ja072316d
(33) Gu,D.;Bongard,H.;Meng,Y.;Miyasaka,K.;Terasaki,O.; Zhang,F.;Deng,Y.;Wu,Z.;Feng,D.;Fang,Y.;Tu,B.; Schüth,F.;Zhao,D.Chem.Mater.2010,22,4828. doi:10.1021/cm101648y
(34) Lu,A.H.;Spliethoff,B.;Schuth,F.Chem.Mater.2008,20, 5314.doi:10.1021/cm800362g
(35) Huang,Y.;Cai,H.;Feng,D.;Gu,D.;Deng,Y.;Tu,B.;Wang, H.;Webley,P.A.;Zhao,D.Chem.Commun.2008,2641. doi:10.1039/b804716b
(36) Liu,F.J.;Li,C.J.;Ren,L.M.;Meng,X.J.;Zhang,H.;Xiao, F.S.J.Mater.Chem.2009,19,7921.doi:10.1039/b910682k
(37) Fang,Y.;Gu,D.;Zou,Y.;Wu,Z.;Li,F.;Che,R.;Deng,Y.;Tu, B.;Zhao,D.Angew.Chem.Int.Edit.2010,49,7987. doi:10.1002/anie.201002849
(38) Sun,Q.;Zhang,X.Q.;Han,F.;Li,W.C.;Lu,A.H.J.Mater. Chem.2012,22,17049.doi:10.1039/c2jm33030j
(39) Liu,D.;Lei,J.H.;Guo,L.P.;Qu,D.Y.;Li,Y.;Su,B.L. Carbon 2012,50,476.doi:10.1016/j.carbon.2011.09.002
(40) Durairaj,R.B.Resorcinol Chemistry,Technology and Applications;Springer-Verlag:New York,2005;pp 179-209.
(41) Al-Muhtaseb,S.A.;Ritter,J.A.Adv.Mater.2003,15,101. doi:10.1002/adma.200390020
(42) Vayssieres,L.;Keis,K.;Hagfeldt,A.;Lindquist,S.E.Chem. Mater.2001,13,4395.doi:10.1021/cm011160s
(43) Liu,D.;Zheng,D.;Wang,L.L.;Qu,D.Y.;Xie,Z.Z.;Lei,J. H.;Guo,L.P.;Deng,B.H.;Xiao,L.;Qu,D.Y.J.Phys.Chem. C 2014,118,2370.doi:10.1021/jp412099y
(44) Liu,D.;Lei,J.H.;Guo,L.P.;Deng,K.J.Carbon 2011,49, 2113.doi:10.1016/j.carbon.2011.01.047
(45) Liu,D.;Zeng,C.;Qu,D.;Tang,H.;Li,Y.;Su,B.L.;Qu,D. J.Power Sources 2016,321,143.doi:10.1016/j. jpowsour.2016.04.129
(46) Wang,J.;Liu,H.Y.;Diao,J.Y.;Gu,X.M.;Wang,H.H.; Rong,J.F.;Zong,B.N.;Su,D.S.J.Mater.Chem.A 2015,3, 2305.doi:10.1039/c4ta05820h
(47) Wang,J.;Liu,H.;Gu,X.;Wang,H.;Su,D.S.Chem. Commun.2014,50,9182.doi:10.1039/c4cc03372h
(48) Wang,G.H.;Cao,Z.;Gu,D.;Pfander,N.;Swertz,A.C.; Spliethoff,B.;Bongard,H.J.;Weidenthaler,C.;Schmidt,W.; Rinaldi,R.;Schuth,F.Angew.Chem.Int.Edit.2016,55,8850. doi:10.1002/anie.201511558
(49) Shen,G.;Sun,X.;Zhang,H.;Liu,Y.;Zhang,J.;Meka,A.; Zhou,L.;Yu,C.J.Mater.Chem.A 2015,3,24041. doi:10.1039/c5ta06129f
(50) Dong,X.;Zhao,X.;Wang,L.;Zhang,M.RSC Adv.2016,6, 48870.doi:10.1039/c6ra06583j
(51) Lu,W.J.;Liu,M.X.;Miao,L.;Zhu,D.Z.;Wang,X.;Duan, H.;Wang,Z.W.;Li,L.C.;Xu,Z.J.;Gan,L.H.;Chen,L.W. Electrochim.Acta 2016,205,132.doi:10.1016/j. electacta.2016.04.114
(52) Liu,D.;Xia,L.J.;Qu,D.Y.;Lei,J.H.;Li,Y.;Su,B.L. J.Mater.Chem.A 2013,1,15447.doi:10.1039/c3ta13518g
(53) Ren,L.;Wu,Q.;Yang,C.;Zhu,L.;Li,C.;Zhang,P.;Zhang, H.;Meng,X.;Xiao,F.S.J.Am.Chem.Soc.2012,134,15173. doi:10.1021/ja3044954
(54) Lin,J.B.;Lin,R.B.;Cheng,X.N.;Zhang,J.P.;Chen,X.M. Chem.Commun.2011,47,9185.doi:10.1039/C1CC12763B
(55) Wang,Q.W.;Mu,Y.J.;Zhang,W.L.;Zhong,L.S.;Meng,Y.; Sun,Y.H.RSC Adv.2014,4,32113.doi:10.1039/c4ra02743d
(56) Zhang,Z.;Wang,B.;Zhu,C.;Gao,P.;Tang,Z.;Sun,N.;Wei, W.;Sun,Y.J.Mater.Chem.A 2015,3,23990.doi:10.1039/ c5ta06465a
(57) Zhu,J.H.;Yang,J.;Miao,R.R.;Yao,Z.Q.;Zhuang,X.D.; Feng,X.L.J.Mater.Chem.A 2016,4,2286.doi:10.1039/ c5ta09073c
(58) Zhang,Z.;Zhu,C.;Sun,N.;Wang,H.;Tang,Z.;Wei,W.;Sun, Y.J.Phys.Chem.C 2015,119,9302.doi:10.1021/acs. jpcc.5b00239
(59) Hu,B.;Wang,K.;Wu,L.;Yu,S.H.;Antonietti,M.;Titirici, M.M.Adv.Mater.2010,22,813.doi:10.1002/ adma.200902812
(60) Titirici,M.M.;White,R.J.;Falco,C.;Sevilla,M.Energy Environ.Sci.2012,5,6796.doi:10.1039/c2ee21166a
(61) Kubo,S.;White,R.J.;Yoshizawa,N.;Antonietti,M.;Titirici, M.M.Chem.Mater.2011,23,4882.doi:10.1021/cm2020077
(62) Feng,S.;Li,W.;Wang,J.;Song,Y.;Elzatahry,A.A.;Xia,Y.; Zhao,D.Nanoscale 2014,6,14657.doi:10.1039/c4nr05629a
(63) Xu,F.;Chen,Y.;Tang,M.;Wang,H.;Deng,J.;Wang,Y.ACS Sustain.Chem.Eng.2016,4,4473.doi:10.1021/ acssuschemeng.6b01196
(64) Schlienger,S.;Graff,A.L.;Celzard,A.;Parmentier,J.Green Chem.2012,14,313.doi:10.1039/c2gc16160e
(65) Braghiroli,F.L.;Fierro,V.;Parmentier,J.;Pasc,A.;Celzard, A.Green Chem.2016,18,3265.doi:10.1039/c5gc02788h
(66) To,J.W.;He,J.;Mei,J.;Haghpanah,R.;Chen,Z.;Kurosawa, T.;Chen,S.;Bae,W.G.;Pan,L.;Tok,J.B.;Wilcox,J.;Bao, Z.J.Am.Chem.Soc.2016,138,1001.doi:10.1021/ jacs.5b11955
(67) Lv,Y.Y.;Zhang,F.;Dou,Y.Q.;Zhai,Y.P.;Wang,J.X.;Liu, H.J.;Xia,Y.Y.;Tu,B.;Zhao,D.Y.J.Mater.Chem.2012,22, 93.doi:10.1039/c1jm12742j
(68) Jin,J.;Tanaka,S.;Egashira,Y.;Nishiyama,N.Carbon 2010, 48,1985.doi:10.1016/j.carbon.2010.02.005
(69) Hao,G.P.;Li,W.C.;Wang,S.A.;Wang,G.H.;Qi,L.;Lu,A. H.Carbon 2011,49,3762.doi:10.1016/j.carbon.2011.05.010
(70) Wang,X.Q.;Liu,C.G.;Neff,D.;Fulvio,P.F.;Mayes,R.T.; Zhamu,A.;Fang,Q.;Chen,G.R.;Meyer,H.M.;Jang,B.Z.; Dai,S.J.Mater.Chem.A 2013,1,7920.doi:10.1039/ c3ta11342f
(71) Wang,X.Q.;Lee,J.S.;Zhu,Q.;Liu,J.;Wang,Y.;Dai,S. Chem.Mater.2010,22,2178.doi:10.1021/cm100139d
(72) Huang,K.;Chai,S.H.;Mayes,R.T.;Tan,S.;Jones,C.W.; Dai,S.Microporous Mesoporous Mat.2016,230,100. doi:10.1016/j.micromeso.2016.04.041
(73) Li,N.W.;Zheng,M.B.;Feng,S.Q.;Lu,H.L.;Zhao,B.; Zheng,J.F.;Zhang,S.T.;Ji,G.B.;Cao,J.M.J.Phys.Chem. C 2013,117,8784.doi:10.1021/jp3127219
(74) Wang,Z.;Stein,A.Chem.Mater.2008,20,1029.doi:10.1021/ cm0717864
(75) Xue,C.F.;Tu,B.;Zhao,D.Y.Nano Res.2009,2,242. doi:10.1007/s12274-009-9022-y
(76) Steinhart,M.;Liang,C.D.;Lynn,G.W.;Gosele,U.;Dai,S. Chem.Mater.2007,19,2383.doi:10.1021/cm070455o
(77) Liu,H.J.;Wang,X.M.;Cui,W.J.;Dou,Y.Q.;Zhao,D.Y.; Xia,Y.Y.J.Mater.Chem.2010,20,4223.doi:10.1039/ b925776d
(78) Petkovich,N.D.;Stein,A.Chem.Soc.Rev.2013,42,3721. doi:10.1039/c2cs35308c
(79) Liu,R.;Shi,Y.;Wan,Y.;Meng,Y.;Zhang,F.;Gu,D.;Chen, Z.;Tu,B.;Zhao,D.J.Am.Chem.Soc.2006,128,11652. doi:10.1021/ja0633518
(80) Tripathi,P.K.;Liu,M.;Zhao,Y.;Ma,X.;Gan,L.;Noonan,O.; Yu,C.J.Mater.Chem.A 2014,2,8534.doi:10.1039/ c4ta00578c
(81) Yu,T.;Deng,Y.H.;Wang,L.;Liu,R.L.;Zhang,L.J.;Tu,B.; Zhao,D.Y.Adv.Mater.2007,19,2301.doi:10.1002/ adma.200700667
(82) Liu,H.J.;Wang,J.;Wang,C.X.;Xia,Y.Y.Adv.Energy Mater. 2011,1,1101.doi:10.1002/aenm.201100255
(83) Liu,D.;Cheng,G.;Zhao,H.;Zeng,C.;Qu,D.Y.;Xiao,L.; Tang,H.L.;Deng,Z.;Li,Y.;Su,B.L.Nano Energy 2016,22, 255.doi:10.1016/j.nanoen.2016.02.022
(84) Cordes,D.B.;Lickiss,P.D.;Rataboul,F.Chem.Rev.2010, 110,2081.doi:10.1021/cr900201r
(85) Podyacheva,O.Y.;Ismagilov,Z.R.Catal.Today 2015,249, 12.doi:10.1016/j.cattod.2014.10.033
(86) Guo,D.;Shibuya,R.;Akiba,C.;Saji,S.;Kondo,T.; Nakamura,J.Science 2016,351,361.doi:10.1126/science. aad0832
(87) Lin,T.;Chen,I.W.;Liu,F.;Yang,C.;Bi,H.;Xu,F.;Huang,F. Science 2015,350,1508.doi:10.1126/science.aab3798
(88) Deng,Y.F.;Xie,Y.;Zou,K.X.;Ji,X.L.J.Mater.Chem.A 2016,4,1144.doi:10.1039/c5ta08620e
(89) Song,J.;Gordin,M.L.;Xu,T.;Chen,S.;Yu,Z.;Sohn,H.;Lu, J.;Ren,Y.;Duan,Y.;Wang,D.Angew.Chem.Int.Edit.2015, 54,4325.doi:10.1002/anie.201411109
(90) Peng,H.J.;Zhang,Q.Angew.Chem.Int.Edit.2015,54, 11018.doi:10.1002/anie.201505444
(91) Shen,W.Z.;Fan,W.B.J.Mater.Chem.A 2013,1,999. doi:10.1039/c2ta00028h
(92) Jurewicz,K.;Babel,K.;Ziolkowski,A.;Wachowska,H. Electrochim.Acta 2003,48,1491.doi:10.1016/S0013-4686 (03)00035-5
(93) Wu,Z.X.;Webley,P.A.;Zhao,D.Y.J.Mater.Chem.2012, 22,11379.doi:10.1039/c2jm16183d
(94) Wei,J.;Zhou,D.;Sun,Z.;Deng,Y.;Xia,Y.;Zhao,D.Adv. Funct.Mater.2013,23,2322.doi:10.1002/adfm.201202764
(95) Song,Y.;Li,L.;Wang,Y.;Wang,C.;Guo,Z.;Xia,Y. ChemPhysChem 2014,15,2084.doi:10.1002/cphc.201402250
(96) Tang,J.;Liu,J.;Li,C.;Li,Y.;Tade,M.O.;Dai,S.;Yamauchi, Y.Angew.Chem.Int.Edit.2015,54,588.doi:10.1002/ anie.201407629
(97) Tang,J.;Liu,J.;Salunkhe,R.R.;Wang,T.;Yamauchi,Y. Chem.Commun.2016,52,505.doi:10.1039/c5cc07610b
(98) Zhu,Y.P.;Liu,Y.;Liu,Y.P.;Ren,T.Z.;Chen,T.;Yuan,Z.Y. ChemCatChem 2015,7,2903.doi:10.1002/cctc.201500148
(99) Deng,C.;Zhong,H.;Li,X.;Yao,L.;Zhang,H.Nanoscale 2016,8,1580.doi:10.1039/c5nr06749a
(100) Zhao,X.;Zhang,Q.;Zhang,B.;Chen,C.M.;Wang,A.; Zhang,T.;Su,D.S.J.Mater.Chem.2012,22,4963. doi:10.1039/c2jm15820e
(101) Kruk,M.Acc.Chem.Res.2012,45,1678.doi:10.1021/ ar200343s
Soft-Templated Ordered Mesoporous Carbon Materials: Synthesis,Structural Modification and Functionalization
LIU Dan HU Yan-Yan ZENG Chao QU De-Yu*
(Department of Chemistry,School of Chemistry,Chemical Engineering and Life Sciences, Wuhan University of Technology,Wuhan 430070,P.R.China)
Ordered mesoporous carbon materials(OMCs)have potentially broad applications in many fields, such as adsorption,separation,catalysis,and energy storage/conversion.Compared with the elaborate hardtemplate strategy,the soft-template approach,which is based on the self-assembly between amphiphilic block copolymers and polymerizable precursors(e.g.,phenolic resins),is a more effective and efficient method for the synthesis of OMCs.In this review,the mechanism and characteristics for three main soft-template methods, i.e.,solvent evaporation-induced self-assembly synthesis,aqueous cooperative self-assembly synthesis and solvent-free synthesis,are discussed and compared.In addition,a few highlights of recent progress,including application of novel carbon precursors,structural modification and functionalization of OMCs,are outlined. Finally,we summarize the crucial issues to be addressed in developing the synthesis methodology of OMCs.
Ordered mesoporous material;Porous carbon;Block copolymer;Soft template; Self-assembly
O647
10.3866/PKU.WHXB201609141
Received:June 24,2016;Revised:September 14,2016;Published online:September 14,2016.
*Corresponding author.Email:deyuquwuhan@163.com;Tel/Fax:+86-27-87756662.
The project was supported by the National Natural Science Foundation of China(21401145,11474226).国家自然科学基金(21401145,11474226)资助项目
猜你喜欢
广州化工(2022年22期)2022-02-01
广州化工(2020年6期)2020-04-18
无机盐工业(2020年11期)2020-01-01
汽车文摘(2018年1期)2018-11-26
环境科技(2017年6期)2018-01-17
无机盐工业(2017年4期)2017-03-10
天然产物研究与开发(2016年6期)2016-06-05
中国塑料(2015年11期)2015-10-14
中国塑料(2015年1期)2015-10-14
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
