
伐地那非的合成工艺改进
2016-12-27 05:13刘志瑞李晓利程卯生
合成化学 2016年12期
刘志瑞,李晓利,吕 佩,程卯生,沙 宇
(沈阳药科大学 基于靶点的药物设计与研究教育部重点实验室,辽宁 沈阳 110016)
·制药技术·
伐地那非的合成工艺改进
刘志瑞,李晓利,吕 佩,程卯生,沙 宇*
(沈阳药科大学 基于靶点的药物设计与研究教育部重点实验室,辽宁 沈阳 110016)
以2-乙氧基苯腈为原料,依次经Pinner反应和肼解反应制得中间体2-乙氧基苯甲亚胺酸酰肼(3);3经两步环合,氯磺化和氨化反应制得伐地那非,其结构经1H NMR,13C NMR和MS(ESI)确证,总收率31.6%。
2-乙氧基苯腈;伐地那非;Pinner反应;药物合成;工艺改进
伐地那非(8),化学名为2-[2-乙氧基-5-(4-乙基-哌嗪-1-磺胺基)-苯基]-5-甲基-7-丙基-3H-咪唑[5,1-f]-[1,2,4]三联氮-4-酮,是德国Bayer公司研发的一种选择性PDE5抑制剂,主要用于治疗男性勃起功能障碍,2003年在美国批准上市[1]。8作为新一代的高选择性PDE5抑制剂具有用量小、起效快、副作用小、持续时间长等优点,现已成为临床治疗男性勃起功能障碍的一线药物[2]。
文献报道了合成8的多种方法[3-6],其中Nowakowski等[3]所报道的方法最常见。即以2-乙氧基苯腈为起始原料,与盐酸羟氨发生加成反应,再经还原、肼解,两步环合、磺化、氯代、胺化等8步反应合成8,总收率25.8%。原路线中Pd/C催化的氢气还原反应需加压设备,故成本较高,而且Pd/C在后处理时易发生火灾,影响产品质量,使其在工业应用上受到限制。并且在合成关键中间体3-正丁酰胺基丁酸乙酯-2-酮(5)时采用一锅法,副产物较多[7,13],产品纯度的稳定性无法保证。因此该路线存在诸多弊端。
本文结合相关文献[7-11],对原路线进行优化改进。以2-乙氧基苯腈(1)为起始原料,经Pinner反应[7,9]合成2-乙氧基苯甲亚胺酸乙酯(2),再进行肼解[7-8],两步环合,氯磺化[10-11],以及胺化等6步反应得到8(Scheme 1),总收率31.6%,其结构经1H NMR,13C NMR和MS(ESI)确证。
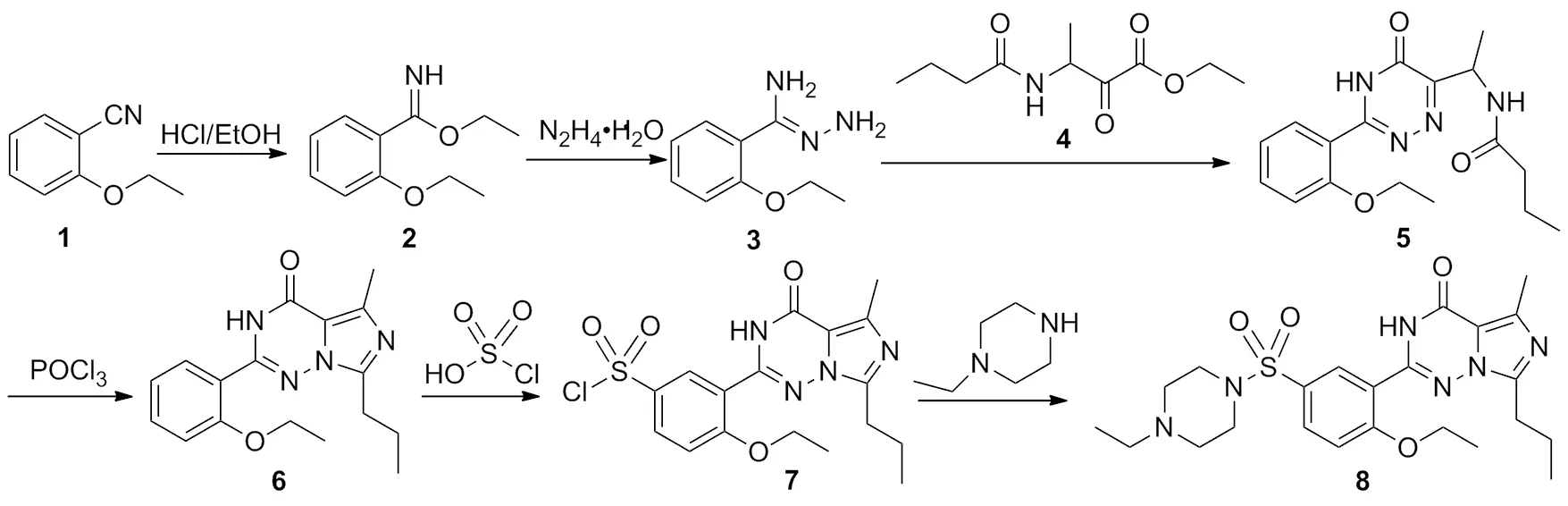
Scheme 1
1 实验部分
1.1 仪器与试剂
B-540型数字熔点仪;Bruker ARX-400 MHz型核磁共振仪(CDCl3为溶剂,TMS内标);Agilent 1260-6120型单四极杆液相色谱质谱联用仪;Waters 2489型液相色谱仪。
1,国药集团;2-乙氧基苯腈(4)按文献[12]方法合成;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1) 2的合成
在三颈瓶中加入1 110.0 g(0.75 mol)和氯化氢的无水乙醇溶液250 mL(4.7 mol·L-1),于室温反应12 h。旋蒸除溶,残余物倒入冰水中,用乙酸乙酯萃取,取水相,于0~5 ℃,向水相缓慢滴加3.0 mol·L-1的氢氧化钠溶液,调至pH>10,用乙酸乙酯(3×500 mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,旋干溶剂得橘黄色固体2 140.9 g,用混合溶剂[V(环己烷):V(乙酸乙酯)=4:1]重结晶得淡黄色固体2 127.3 g,收率87.9%;MS(ESI)m/z:194.2{[M+H]+}。
(2) 2-乙氧基苯甲亚胺酸酰肼(3)的合成
在三颈瓶中加入2 9.7 g(50.0 mmol)和无水甲醇200 mL,于0~5 ℃滴加水合肼2.8 g(55.0 mmol),滴毕,于室温反应12 h。旋蒸除溶,剩余物溶于乙酸乙酯100 mL,用无水硫酸钠干燥过夜。旋蒸除去有机相得淡黄色油状液体3 10.3 g;1H NMRδ:1.43(t,J=7.0 Hz,3H),4.09(q,J=7.0 Hz,2H),4.09(br s,2H),5.14(br,2H),6.90(d,J=8.3 Hz,1H),6.96(dt,J=7.6 Hz,0.84 Hz,1H),7.30(dt,J=8.4 Hz,1.8 Hz,1H),7.74(dd,J=7.7 Hz,1.7 Hz,1H);MS(ESI)m/z:179.1{[M+H]+}。
(3) 5的合成
在三颈瓶中依次加入3 10.3 g(58.0 mmol),4 18.4 g(85.0 mmol)和无水甲醇500 mL,升温回流反应1 h。旋蒸除溶,剩余物加乙酸乙酯,用水洗涤,水相用乙酸乙酯(3×200 mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除溶得橘黄色固体22.1 g,用混合溶剂[V(环己烷):V(丙酮)=4:1]重结晶得淡黄色粉末5 9.7 g,两步总收率58.6%;1H NMRδ:0.85(t,J=7.4 Hz,3H),1.31~1.35(m,6H),1.46~1.55(m,2H),2.04~2.15(m,2H),4.15(q,J=6.9 Hz,2H),5.06(t,J=6.7 Hz,1H),7.10(t,J=7.5 Hz,1H),7.21(d,J=8.4 Hz,1H),7.56(dt,J=7.9 Hz,1.5 Hz,1H),7.65(d,J=7.6 Hz,1H),8.17(d,J=7.6 Hz,1H),13.56(brs,1H);MS(ESI)m/z:331.1{[M+H]+},661.2{[2M+H]+},683.2{[2M+Na]+}。
(4) 2-(2-乙氧基苯基)-5-甲基-7-丙基-3H-咪唑并[5,1-f][1,2,4]三嗪-4-酮(6)的合成
在三颈瓶中加入5 9.7 g(29.3 mmol)和氯仿100 mL,于室温滴加三氯氧磷4.0 mL(44.1 mmol),滴毕,升温回流反应3 h。冷却至室温,将反应液缓慢倒入冰水200 mL中淬灭反应,分液,水相用氯仿(3×200 mL)萃取,合并有机相,依次用饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤至中性,用无水硫酸钠干燥,旋蒸除溶得灰褐色粉末6 8.8 g,收率96.2%;1H NMRδ:0.92(t,J=7.4 Hz,3H),1.31(t,J=6.9 Hz,3H),1.69~1.78(m,2H),2.48(s,3H),2.83(t,J=7.4 Hz,2H),4.11(q,J=6.9 Hz,2H),7.06(d,J=7.4 Hz,1H),7.16(d,J=8.1 Hz,1H),7.49~7.54(m,2H),11.50(s,1H);MS(ESI)m/z:313.1{[M+H]+};GC-MSm/z:312.2,297.2,284.2, 269.2,256.2,166.2,67.1。
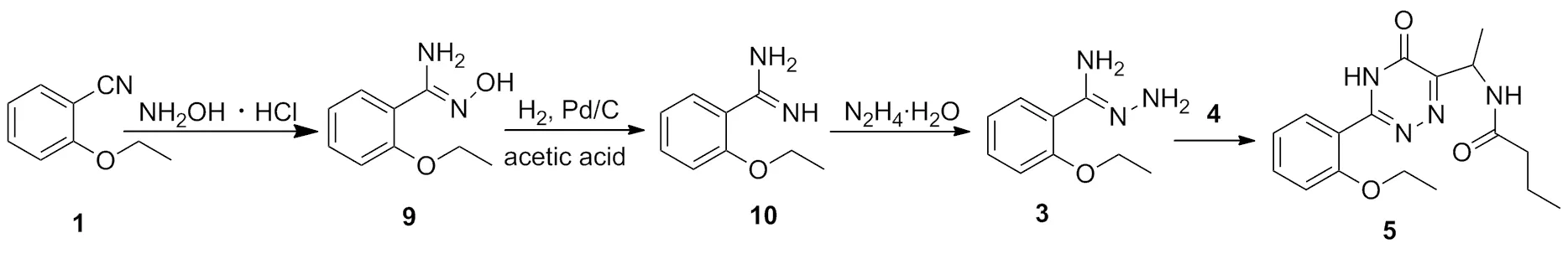
Scheme 2
(5) 2-(2-乙氧基-5-氯磺酰基苯基)-5-甲基-7-丙基-3H-咪唑并[5,1-f][1,2,4]三嗪-4-酮(7)的合成
在三颈瓶中加入6 8.8 g(28.2 mmol)和氯仿500 mL,滴加氯磺酸5.8 mL(85.0 mmol),滴毕,升温回流反应1 h。冷却至室温,将反应液缓慢滴入冰水中淬灭反应,分液。水相用氯仿(3×200 mL)萃取,合并有机相,依次用饱和碳酸氢钠溶液与饱和氯化钠溶液洗涤至中性,用无水硫酸钠干燥,旋蒸除溶得淡黄色粉末7 10.4 g,收率88.7%;MS(ESI)m/z:411.1{[M+H]+}。
(6) 8的合成
在三颈瓶中加入7 10.4 g(25.3 mmol),三乙胺3.8 g(38.0 mmol)和氯仿100 mL,搅拌下滴加1-乙基哌嗪4.8 mL(38.0 mmol),滴毕,反应30 min。有机相用饱和碳酸氢钠溶液与饱和氯化钠溶液洗涤至中性,用无水硫酸钠干燥,用混合溶剂[V(石油醚):V(乙酸乙酯)=5:1]重结晶得类白色晶体8.9 g,收率72.0%,纯度99.6%[HPLC(流动相:水/乙腈=65/35,V/V),pH=6.0,面积归一化法],m.p.191.0~191.7 ℃;1H NMRδ:1.02(s,3H),1.08(t,J=7.3 Hz,3H),1.60(t,J=7.0 Hz,3H),1.82~1.91(m,2H),2.48(br s,2H),2.61(br s,4H),2.64(s,3H),2.99(t,J=7.7 Hz,2H),3.14(br s,4H),4.34(q,J=7.0 Hz,2H),7.16(d,J=8.8 Hz,1H),7.88(dd,J=8.8 Hz,2.3 Hz,1H),8.45(d,J=2.3 Hz,1H),9.68(s,1H);13C NMRδ:160.7,155.5,146.6,144.8,138.1,132.6,130.6,126.4,121.4,114.1,113.7,65.4,51.7,51.5,46.4,27.6,20.7,14.7,14.6,14.2,12.3;MS(ESI)m/z:245.1{[M+2H]+},489.2{[M+H]+},977.3{[2M+H]+};HR-MS(ESI)m/z:489.230 4{[M+H]+}。
2 结果与讨论
2.1 合成
(1) 5的合成
5是合成8的关键中间体。文献[13]报道的合成方法以1为起始原料,首先与盐酸羟胺加成生成2-乙氧基-N-羟基苯甲咪(9),然后9在钯碳催化下经氢气还原生成2-乙氧基苯甲咪(10);10再与水合肼发生肼解反应生成3;3与中间体4环合生成5(Scheme 2)。
该路线中从化合物1制备5需要四步反应,从10到5用一锅法制备,不仅收率低而且副反应较多。本文由1通过Pinner反应制备2,纯化后的2与水合肼几乎是定量反应生成3;3与4反应制备5,化合物1经三步反应制备化合物5。综上所述,该文所用路线较短,且产品纯度高,适合大规模生产。
(2) 8的合成
原路线中,6首先经浓硫酸磺化,然后与氯化亚砜反应生成7;7与1-乙基哌嗪胺化生成8,中间经历了三步反应,无形中增加了设备成本和时间成本。本文所用路线直接用6与氯磺酸反应,一步生成7;7与1-乙基哌嗪胺化生成8。这样本文路线由6合成8减少为两步反应,收率相当,但产品质量得到提高,生产成本降低。
对伐地那非的合成工艺进行了改进。以2-乙氧基苯腈为起始原料,经过Pinner反应、肼解、两步环合、氯磺化、以及胺化等6步反应合成伐地那非。本文所用路线较短,原料廉价易得,反应条件温和,副反应少,后处理简单,总收率为31.8%,高于文献[2]报道的25.8%。产品纯度大于99.6%,单杂均小于0.1%,符合中国药典对原料药的质量要求。
[1] Sorbera L A,Martín L,Rabasseda X,etal.Treatment of erectile dysfunction phosphodiesterase 5 Inhibitor[J].Drugs of the Future,2001,26(2):141-144.
[2] Dunn P J,Synthesis of commercial phosphodiesterase(V) inhibitors[J].Organic Process Research &Development.2005,9(1):88-97.
[3] Nowakowski M,Gehring R,Heilman W,etal.Preparation of 2-(sulfonamidophenyl)imidazotriazinones:WO 200250076[P].2002.
[4] Teresa O,Ewa P,Gajewska,etal.Alternative method for the synthesis of imidazo[5,1-f][1,2,4]triazin-4(3H)-one——a substrate for the preparation of phosphodiesterase (5) inhibitors[J].Tetrahedron,2013,69(2):474-480.
[5] Helmut H,Ulrich N,Thomas Schenke,etal.Imidazo[5,1-f][1,2,4]triazin-4(3H)-ones,a new class ofpotent PDE-5 inhibitors[J].Bioorganic &Medicinal Chemistry Letters,2002,33(30):865-868.
[6] Alexander H R,Jason H.A novel method for the synthesis of imidazo[5,1-f][1,2,4]-triazin-4(3H)-ones[J].J Org Chem,2005,70(18):7331-7337.
[7] Wamhoff H,Tzanova M.Novel 6-azapteridines from bifunctional 1,2,4-triazines[J].Collect Czech Chem Commun,2003,34(34):965-974.
[8] Dward C,Taylor,Stephen F,etal.Synthesis of some 7-aryl-6-azapteridines from 1,2,4-triazine intermediates[J].J Org Chem,1972,37(24):1972-3959.
[9] Olivier B,Sharon W,Jean F D,etal.Reverse-benzamidine antimalarial agents:Design,synthesis,and biological evaluation[J].Bioorganic &Medicinal Chemistry Letters,2010,20(19):5815-5817.
[10] Detlef G,Raafat S,Farid S,etal.Synthesis of new series of pyrazolo[4,3-d]pyrimidin-7-ones,and pyrido[2,3-d]pyrimidin-4-ones for their bacterial and cyclin-dependent kinases (CDKs) inhibitory activities[J].Med Chem Res,2010,20(4):408-420.
[11] Hyun S,Lee J Y,Dae K K.Synthesis of 5-ethyl-2-{5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl}-7-n-propyl-3,5-dihydro-4H-pyrrolo[3,2-d]-[2-14C]pyrimidin-4-one.2HCl(14C-SK3530·2HCl)[J].J Label Compd Radiopharm,2006,49(13):1141-1149.
[12] Zhao Z L,Anne C O V,Girish S,etal.Novel peptidylα-keto amide inhibitors of calpains and other cysteine proteases[J].J Med Chem,1996,39(20):4089-4098.
[13] Isabel C,David W,Latham,etal.Application of the dakin-west reaction to the synthesis of imidazo -[5,1-f]-1,2,4-triazin-4(3H)-ones[J].J Chem Soc,1980,11(27):1139-1146.
Process Improvement on the Synthesis of Vardenafil
LIU Zhi-rui,LI Xiao-li,LÜ Pei,CHENG Mao-sheng,SHA Yu*
(Key Laboratory of Structure-Based Drug Design &Discovery,Ministry of Education, Shenyang Pharmaceutical University,Shenyang 110016,China)
An intermediate,2-ethoxybenzohydrazonamide(3),was prepared by the reaction of Pinner and hydrazinolysis from 2-ethoxybenzonitrile.Vardenafil was synthesized by two successive cyclizations,chlorosulfonation,and amination from 3.The structure was confirmed by1H NMR,13C NMR and MS(ESI).The total yield was 31.6%.
2-ethoxybenzonitrile;vardenafil;Pinner reaction;drug synthesis;process improvement
2016-04-19;修改日期:2016-09-16
刘志瑞(1988-),男,汉族,山东临沂人,硕士研究生,主要从事化学原料药和医药中间体的合成工艺研究。 E-mail:liuzhiruirui@163.com
沙宇,副教授,Tel.024-23986418,E-mail:shayu@syphu.edu.cn
R914.5;O621.3
A
10.15952/j.cnki.cjsc.1005-1511.2016.12.16108
猜你喜欢
长江大学学报(自科版)(2021年3期)2021-06-01
药学与临床研究(2020年3期)2020-07-11
理化检验-化学分册(2020年5期)2020-06-15
电脑报(2020年10期)2020-04-28
硫酸工业(2020年2期)2020-04-16
铜仁学院学报(2018年6期)2018-07-05
文物保护与考古科学(2016年1期)2016-04-16
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
