
基于α调整的国际多中心临床试验目标区二分类终点有效性研究的桥接方法*
2016-12-27 08:49:07第四军医大学军事预防医学院卫生统计学教研室710032
中国卫生统计 2016年1期
第四军医大学军事预防医学院卫生统计学教研室(710032)
虞成凯 王 陵△ 戚洋洋 李婵娟 李 晨 蒋志伟 夏结来△
基于α调整的国际多中心临床试验目标区二分类终点有效性研究的桥接方法*
第四军医大学军事预防医学院卫生统计学教研室(710032)
虞成凯 王 陵△戚洋洋 李婵娟 李 晨 蒋志伟 夏结来△
目的 探索采用合理的调整后检验水准α′作为国际多中心临床试验(MRCT)在目标区的决策依据的可行性,为MRCT在目标区通过审批提供参考。方法 利用Monte Carlo模拟构建二分类资料、优效性设计、包含目标区的MRCT数据模型,比较不同规模的MRCT在检验水准α=0.05水平显示试验组优效的前提下,目标区采用调整后检验水准α′作为决策依据的条件Ⅰ型错误率(CFPR)、条件检验效能(CP)随目标区样本量占总样本量比例K的变化情况。结果 在不同规模的MRCT中,目标区的CFPR和CP均随着检验水准α′、样本比例K的增加而增加;当α′=0.5时,CFPR和CFPR′基本可以被控制在50%和5%左右。当样本比例K固定时,目标区CP相对稳定,尤其当K≥30%时随着α′的增加目标区CP几乎不受MRCT总样本量N的影响,且当α′不低于0.3时基本可以确保足够的CP(≥80%)。当K≥30%且设置α′=0.5时,即使目标区试验药物疗效略低于MRCT的疗效即f≥0.8,也可以确保目标区足够的CP和CP′(≥80%)。结论 建议MRCT在方案设计时,目标区的样本比例不应低于30%,试验数据在目标区的检验水平不能超过0.5。该标准适用于目标区试验药物疗效略低于MRCT的疗效即f≥0.8的情形,若估计目标区试验药物疗效与MRCT的疗效相当,检验水平可适当降低,但不建议低于0.3。
条件Ⅰ型错误率 条件检验效能 国际多中心临床试验 Monte Carlo模拟
当研究药物在原地区批准上市后,申办者可能会在一个新的地区申请注册该药物。然而,原地区和新地区的种族、文化和临床实践之间的差异及其对药物安全性、有效性、剂量和给药方案的影响,限制了新地区药物监管机构接受原地区产生的临床试验数据的意愿。因此新地区药物监管机构通常要求申办者重复类似试验,以桥接研究药物在本地区的有效性和安全性。但是,在新地区的重复临床评估不仅需要昂贵的研发资源而且大大延迟了新地区患者获得新药的时间[1]。为了解决这种困境,人用药品注册技术要求国际协调会(ICH)E5指南[2],即《接受国外临床资料的种族影响因素》给出了基于桥接策略评价种族因素对研究药物有效性和安全性影响的框架[3]。然而,大部分桥接试验是在研究药物经原地区批准上市后才在新地区进行,同样存在新药上市滞后的问题。
为了加快研究药物在日本上市,日本厚生劳动省2007年发布的指南《全球临床试验的基本原则》[4]用问答的形式列出了计划和实施国际多中心临床试验(multi-regional clinical trial,MRCT)的基本概念,提出在MRCT中应纳入足够多的日本病例来评价潜在的种族差异以确保新药能够在日本同步上市。该原则列举了两种估计日本地区样本量的方法。Quan[5]等人讨论了方法一,提出日本区纳入的样本量须保证本地区试验组与对照组疗效差值大于或等于整个试验疗效差值的50%的概率不低于0.8。Chen[6]等人在Quan的研究基础上提出当MRCT在检验水准α=0.05的水平显示试验组优效于对照组时,可采用调整后检验水准α′作为局部地区的药物监管部门批准研究药物上市的决策依据。本研究将进一步研讨二分类资料的MRCT采用检验水准α′作为目标区决策依据的可行性并探索α′的合理取值。
方法与步骤
假定一个二分类资料、优效性设计的MRCT,包括目标区和非目标区,试验组与对照组例数比1:1,在总的检验水准α=0.05水平,试验组的疗效优于对照组且差别有统计学意义。
采用Monte Carlo模拟构建MRCT数据模型,比较不同规模的MRCT中目标区采用不同检验水准α′作为决策依据允许研究药物上市的条件Ⅰ型错误率(conditional false positive rate,CFPR)和条件检验效能(conditional test power,CP)随目标区样本量占总样本量比例的变化情况。本研究所有的数据均由模拟产生,采用SAS 9.1.3统计分析软件编写程序和统计分析。
1.参数设置
令MRCT的试验组与对照组疗效分别为Pt和Pr,其中目标区的试验组与对照组疗效分别为PtL和PrL,非目标区的试验组与对照组疗效分别为PtNL和PrNL,假定 Pr=PrL=PrNL,Pt=PtNL。
参考 Huang[7]等人研究的参数设置,设 Pr为0.20,为了获得大、中、小规模的样本量,令试验组与对照组效应差δ分别为0.05,0.08,0.20,则对应的Pt分别为0.25,0.28,0.40,采用 PASS11.0软件计算相应的样本量N分别为1094,447,82。设目标区样本量占总样本量的比例K分别为10%,30%,50%,调整后的检验水准 α′分别设为0.1,0.2,…,0.9。模拟研究包括以下三个部分:①当PtL=Pr,即PtL=0.20时计算并比较目标区的CFPR;②当PtL=Pt时计算并比较目标区CP;③当PtL略低于Pt,即目标区试验组与对照组效应差δL为 δ的f倍(即 PtL=f×δ+Pr)时计算并比较目标区 CP,其中 f设为 0.5,0.6,0.7,0.8。
2.模拟步骤
(1)根据预先设定的参数,调用SAS均匀分布随机函数RANUNI(seed)分别生成4个包含随机数Xi(0~1)和指示变量 Yi(0,1)的数据集(i=1,2,3,4),分别代表目标区试验组(Y1=1|X1≤PtL,Y1=0|X1>PtL)、对照组(Y2=1|X2≤Pr,Y2=0|X2>Pr)和非目标区试验组(Y3=1|X3≤Pt,Y3=0|X3>Pt)、对照组(Y4=1|X4≤Pr,Y4=0|X4>Pr)的临床试验数据。
2)将4个数据集全部合并作为MRCT的模拟数据,进行两样本率组间比较的卡方检验,P≤0.05认为有统计学差异。
3)在步骤2)的卡方检验显示P≤0.05且^Pt>^Pr的前提下,将目标区试验组与对照组数据集合并作为目标区的模拟数据,进行两样本率组间比较的卡方检验,PL≤α′认为有统计学差异。
4)重复步骤1)~3),循环10000次,计算PL≤α′且^PtL>^PrL的例数nL占P≤0.05且^Pt>^Pr的例数n的百分比。
以上四个步骤基于PtL=Pr时计算目标区CFPR,基于Pr<PtL≤Pt时计算目标区CP。
模拟结果
模拟研究①:整体来看,假定PtL=Pr时,目标区的CFPR随着总样本量N、目标区样本比例K和检验水准α′的增加而增加(图1),且这一变化趋势在大样本的MRCT中趋于稳定。当检验水准α′不超过0.5时基本可以控制CFPR在50%左右。
模拟研究②:当PtL=Pt时,目标区的CP随着目标区样本比例K和检验水准α′的增加而增加,当α′较大时CP的增幅逐渐平缓,且这一变化趋势在大样本的MRCT中趋于稳定(图2)。值得注意的是,当K固定时,目标区的CP相对稳定,尤其当K≥30%时,随着α′的增加目标区CP几乎不受总样本量N的影响,且当α′不低于0.3时基本可以确保足够的 CP(≥80%)。
考虑到目标区与非目标区之间人种的差异,研究药物在目标区的疗效与MRCT疗效并不一定相等,基于模拟研究①和②,暂定α′为0.5,进一步研究目标区试验组疗效略低于MRCT试验组疗效时目标区CP情况。
模拟研究③:当 PtL=f×δ+Pr且 α′=0.5时,目标区的CP随着f和K的增加而增加(表1),当K固定时,CP相对稳定,几乎不受总样本量的影响。相对K而言,f对CP的影响并不明显。当K=30%、f=0.8时,基本可以确保足够的CP(≥80%)。
目标区样本比例K的确定
考虑到本研究模拟背景是在MRCT显示优效的前提下目标区实际无效(概率为100%)时的CFPR、以及目标区实际优效(概率为100%)时的CP,这与现实显然是不符的。此处给一个符合常理的假定:MRCT显示优效的研究药物中,有90%在目标区也是优效的,10%在目标区无效。记:试验药物在目标区无效;:试验药物在目标区优效;:MRCT试验药物无效;:MRCT试验药物优效。
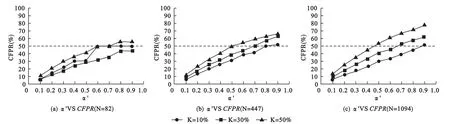
图1 总样本量N、目标区样本比例K、检验水准α′与CFPR的变化关系
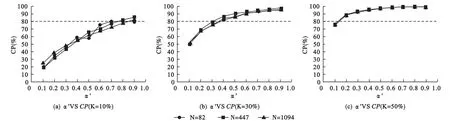
图2 总样本量N、目标区样本比例K、检验水准α′与CP的变化关系

表1 目标区试验组疗效比MRCT试验组疗效略低时的CP(%)

在α′=0.5的情况下,保证K在50%以内,基本可以控制 CFPR(<50%)和 CFPR′(<5%);当 K=10%时,即使 PtL=Pt,依然不能获得预期的 CP(<70%)和CP′(<63%);当K=30%且 f≥0.8时,可以确保足够的CP(≥80.4%)和CP′(≥72.4%);当K=30%时,不同f对应的目标区CFPR′以及CP′见表2。

表2 当K=30%时,不同f对应的目标区CFPR′以及CP′
此外,K=30%作为MRCT目标区的样本比例,具有较好的理论解释。根据两样本率组间比较卡方检验的样本量计算公式[8](公式(1)),本研究 α=0.05,α′=0.5且 1-β=1-β′=0.8,分别计算样本量后作两者的比值(公式(2)),得到目标区样本量占总样本量的比例近似为30%,即目标区试验组疗效与MRCT的试验组疗效相等且K=30%时,在α′=0.5的检验水准下,有80%的检验效能能检验出目标区试验组优于对照组且差别有统计学意义。
讨 论
国际多中心临床试验为同步评价研究药物对全球各地区患者的有效性、安全性提供了可能,然而,它也给各地区的药物监管部门带来了挑战,即如何合理运用MRCT中局部区(例如目标区)的试验数据来制定相关决策依据使得研究药物在本地区能同步上市[9]。Chen等人同时研究了日本提出的方法一和局部区采用调整后检验水准α′的方法,给出了这两种决策依据样本量的计算以及它们之间关系[6]。然而,对于局部区的CFPR并未给出更好的控制建议和相关标准的具体意见。
本研究通过Monte Carlo模拟,在总的检验水准α=0.05的水平试验组的疗效优于对照组且差别有统计学意义的前提下,比较MRCT中目标区设置不同的α′对研究药物在本地区的 CFPR、CFPR′、CP和 CP′的影响。研究发现①当α′≤0.5时能够有效地控制CFPR′在0.05以内;②当K≥30%时,若目标区试验药物疗效与MRCT的疗效相当,设置α′≥0.3可以确保目标区足够的CP(≥80%);③当K≥30%且设置α′=0.5时,即使目标区试验药物疗效略低于MRCT的疗效即f≥0.8,也可以确保目标区足够的CP和CP′(≥80%)。
综上,建议MRCT在方案设计时目标区的样本比例不应低于30%、检验水平不能超过0.5。该标准适用于目标区试验药物疗效略低于MRCT的疗效即f≥0.8的情形,若估计目标区试验药物疗效与MRCT的疗效相当,检验水平可适当降低,但不建议低于0.3。若估计目标区试验药物疗效远低于MRCT的疗效,例如f<0.8,应适当增大目标区的样本比例,例如K>50%,然而一般情况下这一比例并不切实际。
[1]Liu JP,Chow SC.Bridging studies in clinical development.J Biopharm Stat,2002,12(3):359-367.
[2]International Conference on Harmonisation;guidance on ethnic factors in the acceptability of foreign clinical data;availability-FDA.Notice.Fed Regist,1998,63(111):31790-31796.
[3]Ando Y,Hamasaki T.Practical issues and lessons learned from multiregional clinical trials via case examples:a Japanese perspective.Pharm Stat,2010,9(3):190-200.
[4] Ministry Of Health LAWO and Japan,Basic Principles on Global Clinical Trials.2007.
[5]Quan H,et al.Sample size considerations for Japanese patients in a multi-regional trial based on MHLW guidance.Pharm Stat,2010,9(2):100-112.
[6]Chen X,et al.Decision rules and associated sample size planning for regional approval utilizing multiregional clinical trials.J Biopharm Stat,2012,22(5):1001-118.
[7]Huang Q,et al.Design and sample size considerations for simultaneous global drug development program.JBiopharm Stat,2012,22(5):1060-1073.
[8]Shein-Chung Chow.Sample Size Calculations in Clinical Research.Second Edition.2008,Chapman&Hall/CRC.
[9]Shih WJ.Clinical trials for drug registrations in Asian-Pacific countries:proposal for a new paradigm from a statistical perspective.Control Clin Trials,2001,22(4):357-366.
A Bridging Method Based onαAdjustment to Evaluate the Effectiveness of MRCT in Target Region for Dichotomous Endpoint
Yu Chengkai,Wang Lin,Qi Yangyang,et al
(Department of Health Statistics,Faculty of Preventive Medicine,Fourth Military Medical University(710032),Xi′an)
Objective To explore the feasibility of adopting rational adjusted significance level as the decision rule in the target region in MRCT and make reference for evaluating new drug application of MRCT in target region.Methods By Monte Carlo simulation,we studied the conditional false positive rate(CFPR)and conditional test power(CP)changing with the proportions of the sample size in target region by adopting different significance levels given that the treatment effect is shown to be significant under the significance levelα=0.05 based on overall MRCT data for dichotomous data.Results The CFPR and CP are increasing with the significance levelα′and the ratio of sample size K increasing in target region.When the significance levelα′=0.5 the CFPR and CFPR′can be controlled under the 50%and 5%in basic.When the ratio of sample size is fixed,the CP in target region is relatively stable and can′t be influenced by the sample size of local region.Especially when K≥30%,the CP in target region is almost not affected by the total sample size.Whenα′isn′t lower than 0.3,we can ensure enough CP(≥80%).Conclusion When protocol is designed,it is advisable that the ratio of sample size K shouldn′t be less than 30%.The significance levelα′in target region shouldn′t exceed 0.5.The decisional rule is suitable for the difference between the trial and the control group in target region is more than 0.8 times the difference in MRCT.If the estimated treatment effect of the drug in target region is almost like it in MRCT,the significance level can be lower,however,less than 0.3 is not recommended.
Conditional false positive rate;Conditional test power;Multi-regional clinical trial;Monte Carlo simulation
*:国家自然科学基金资助(项目编号:81273176,81302509,81473069)
△通信作者:夏结来,E-mail:jielaixia@yahoo.com;王陵,E-mail:wl.medstat@gmail.com
郭海强)
猜你喜欢
——以三江口港产城新区为例
华南地震(2022年1期)2022-04-06 05:54:22
内蒙古统计(2021年4期)2021-12-06 02:49:20
北京测绘(2021年1期)2021-02-05 11:43:46
——以河南省为例
办公室业务(2019年14期)2019-08-01 02:33:28
家庭影院技术(2018年11期)2019-01-21 02:20:42
测控技术(2018年4期)2018-11-25 09:46:52
上海精神医学(2017年5期)2017-11-29 06:03:10
山东工业技术(2015年13期)2015-07-27 05:23:43
测绘科学与工程(2013年6期)2013-03-11 15:07:54
统计与决策(2012年14期)2012-07-25 08:15:34
