
Faecalibacteriumprausnitzii对实验性结肠炎中TLR4/NF-κB信号通路的影响*
2016-12-23 07:52陈兆桂于成功
胃肠病学 2016年11期
陈兆桂 于成功
南京大学医学院附属鼓楼医院消化科1(210008) 南京鼓楼医院集团仪征医院消化科2
·论 著·
Faecalibacteriumprausnitzii对实验性结肠炎中TLR4/NF-κB信号通路的影响*
陈兆桂1#于成功1,2&
南京大学医学院附属鼓楼医院消化科1(210008) 南京鼓楼医院集团仪征医院消化科2
背景:Faecalibacteriumprausnitzii(Fp)是一种肠道共生菌,其数量减少是炎症性肠病(IBD)患者肠道菌群紊乱的重要特征之一。既往研究显示Fp上清液在实验性结肠炎中具有明显的抗炎效应。目的:探讨Fp上清液在实验性结肠炎中的抗炎效应是否与抑制TLR4/NF-κB信号通路激活有关。方法:24只C57BL/6小鼠随机分为正常对照组、结肠炎模型组和Fp上清治疗组。通过予小鼠连续5 d饮用3.5%葡聚糖硫酸钠(DSS)建立急性结肠炎模型,观察小鼠体质量和结肠组织病理学变化,分别以real-time PCR和免疫组化法检测结肠组织TLR4 mRNA和磷酸化NF-κB p65(p-NF-κB p65)表达,ELISA法检测血清肿瘤坏死因子-α(TNF-α)、白细胞介素-6(IL-6)、环氧合酶-2(COX-2)水平。结果:与正常对照组相比,结肠炎模型组小鼠体质量明显下降,结肠缩短,结肠组织病理学评分、TLR4 mRNA和p-NF-κB p65表达以及血清TNF-α、IL-6、COX-2水平均显著升高(P<0.05)。Fp上清治疗组上述指标均较结肠炎模型组显著改善(P<0.05),其中血清炎症因子水平与正常对照组相比无明显差异(P>0.05)。结论:Fp上清液通过抑制TLR4/NF-κB信号通路激活及其下游炎症因子表达,在实验性结肠炎中发挥抗炎效应。
Faecalibacteriumprausnitzii; 结肠炎; Toll样受体4; NF-κB; 肿瘤坏死因子α; 白细胞介素6; 环氧合酶2
炎症性肠病(inflammatory bowel disease, IBD)是一种在全球范围内广为流行的慢性肠道炎症性疾病,其发病与免疫、环境、遗传等多种因素有关,近年来肠道菌群紊乱在IBD发病中的作用受到广泛关注。Faecalibacteriumprausnitzii(F.prausnitzii, Fp)是一种肠道共生菌,主要定植于末端回肠和回盲部,参与维持肠道健康并为肠道细胞提供能量,Fp数量异常是IBD患者肠道菌群紊乱的重要特征之一[1]。研究发现溃疡性结肠炎(UC)患者肠道内Fp数量的恢复有利于维持临床缓解,而复发者Fp数量相对较低[2];CD患者中,Fp数量减少与高术后复发风险相关[3]。既往研究[3-4]显示,Fp上清液在实验性结肠炎中具有明显的抗炎效应,其机制可能与上调外周血和脾脏中的调节性T细胞(Treg细胞)以及阻断NF-κB炎症信号通路激活、抑制促炎细胞因子分泌有关。肠道炎症信号通路复杂多样,NF-κB是其中的关键环节,而Toll样受体4(Toll-like receptor 4, TLR4)则是NF-κB信号的上游分子之一[5]。为此,本实验从TLR4/NF-κB信号通路角度入手,探讨Fp上清液在实验性结肠炎中的抗炎机制,以期为IBD的治疗提供新策略。
材料与方法
一、实验动物、菌株和主要试剂
雌性SPF级C57BL/6小鼠24只,体质量18~22 g,由南京大学医学院附属鼓楼医院实验动物中心提供;Fp(ATCC 27766,美国模式菌种保藏中心)。高 纯度葡聚糖硫酸钠(DSS)(MP Biomedicals, LLC);RNAiso Plus总RNA提取试剂、PrimeScriptTM逆转录试剂盒(Perfect Real Time)、SYBR®Premix Ex TaqTMⅡ(Tli RNaseH Plus)(TAKARA BIO INC.),PCR引物由生工生物工程(上海)股份有限公司合成;兔抗小鼠磷酸化NF-κB p65(p-NF-κB p65)多克隆抗体(Santa Cruz Biotechnology, Inc.);小鼠肿瘤坏死因子-α(TNF-α)、白细胞介素-6(IL-6)、环氧合酶-2(COX-2)ELISA试剂盒(江苏凯基生物技术股份有限公司)。
二、方法
1.Fp上清液制备:Fp活菌冻干粉解冻,接种至改良培养基,37 ℃厌氧箱中培养。收集对数生长末期细菌培养液,离心,收集上清液冻干,100 mL上清液约可得到3.5 g 冻干粉,分装后-80 ℃保存。使用前每3.5 g冻干粉溶解于20 mL蒸馏水中,4 ℃保存备用。
2.结肠炎模型建立和动物分组干预:24只小鼠随机分为正常对照组、结肠炎模型组和Fp上清治疗组,每组8只,称重并编号。实验第1~5 d,结肠炎模型组和Fp上清治疗组小鼠自由饮用含 3.5% DSS的蒸馏水以建立急性结肠炎模型,正常对照组小鼠自由饮用蒸馏水;Fp上清治疗组小鼠实验第1~8 d每天予0.1 mL/mg Fp上清液灌胃。实验过程中每日观察大鼠精神状态、饮食、饮水、排便情况和体质量变化。实验第8 d,各组小鼠摘眼球取血,离心取上清液,-80 ℃保存,用于ELISA检测;断颈处死小鼠,剖腹取全结肠,测量并记录结肠长度,留取结肠组织(去除回盲部后的前半段结肠),用于HE染色、免疫组化染色和real-time PCR。
3.组织病理学检查:结肠组织4%甲醛溶液固定,石蜡包埋、切片,HE染色,光学显微镜下观察,参照文献报道的方法行组织病理学评分[6]。0分:无炎症证据;1分:低度炎症,伴散在的单核细胞浸润(1~2个浸润灶);2分:中等程度炎症,伴多个浸润灶;3分:高度炎症,血管密度增高,结肠壁增厚;4分:最高度炎症,透壁性炎症细胞浸润,杯状细胞减少。
4. Real-time PCR:取结肠组织,以RNAiso Plus试剂提取总RNA,逆转录合成cDNA,行real-time PCR扩增,操作步骤参照试剂盒说明书。PCR引物序列:TLR4 F 5’-CAA GAA CAT AGA TCT GAG CTT CAA CCC-3’, R 5’-GCT GTC CAA TAG GGA AGC TTT CTA GAG-3’; β-actin F 5’-TGG TAC CAC CAT GTA CCC AG-3’, R 5’-AAG GGT GTA AAA CGC AGC TC-3’。2-△△Ct法计算TLR4 mRNA相对表达量。
5. 免疫组化染色:采用Elivison二步法。结肠组织切片常规脱蜡至水, 3% H2O2灭活内源性过氧化物酶,抗原热修复,山羊血清封闭20 min,依次加入兔抗小鼠 p-NF-κB p65抗体(1∶400)和二抗,DAB显色,苏木精复染,中性树胶封片,光学显微镜下双盲法阅片。
免疫组化评分标准:细胞核中出现棕褐色颗粒为阳性细胞。于高倍镜下随机选取5个视野,计算一个高倍视野下不同着色强度评分(无,0分;弱,1分;中,2分;强,3分)与该着色强度下阳性细胞百分率评分(<5%,0分;5%~9%,1分;10%~19%,2分;20%~50%,3分;≥50%,4分)的乘积,得到的4个乘积相加,即为该高倍视野下的免疫组化评分。同样方法计算另4个高倍视野下的免疫组化评分,总分为5个视野评分之和。
6.ELISA:取大鼠血清,参照 ELISA试剂盒说明书检测TNF-α、IL-6、COX-2水平。
三、统计学分析
结 果
一、一般情况
正常对照组小鼠实验过程中精神状态良好,饮食、饮水正常,粪便成形,体质量未见下降。实验第4d,结肠炎模型组和Fp上清治疗组小鼠出现精神萎靡、饮食、饮水减少、稀便和体质量下降,Fp上清治疗组症状稍轻;第5d,两组均可见肉眼血便,体质量继续下降;第6d,结肠炎模型组2只大鼠死亡,死因为中毒性巨结肠和肠梗阻。死亡大鼠不纳入后续各项指标的统计学分析。
比较实验过程中各组小鼠体质量占原始体质量的百分率,结果显示与正常对照组相比,结肠炎模型组和Fp上清治疗组小鼠体质量下降明显,Fp上清治疗组降幅低于结肠炎模型组(图1A)。与正常对照组相比,结肠炎模型组和Fp上清治疗组小鼠结肠显著缩短(P<0.01),其中结肠炎模型组又短于Fp上清治疗组(P<0.05)(图1B)。
二、结肠组织病理学变化
正常对照组小鼠结肠黏膜上皮完整,无充血水肿,腺体排列整齐;结肠炎模型组小鼠结肠黏膜可见不同程度的充血水肿,黏膜上皮破坏,有典型溃疡形成,镜下见大量炎性细胞浸润,黏膜和黏膜下层血管密度增高,腺体结构紊乱;Fp上清治疗组小鼠结肠黏膜稍有充血水肿,镜下见少量炎性细胞浸润,血管增生不明显,腺体结构较结肠炎模型组完整清晰(图2A-C)。结肠炎模型组结肠组织病理学评分显著高于正常对照组(P<0.001),Fp上清治疗组评分较结肠炎模型组降低(P<0.05),但仍显著高于正常对照组(P<0.001)(图2D)。
三、结肠组织TLR4mRNA表达
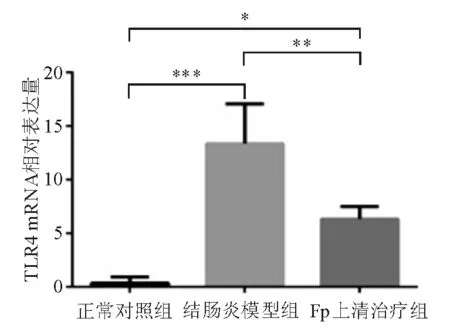
Real-timePCR检测显示,结肠炎模型组结肠组织TLR4mRNA相对表达量显著高于正常对照组(P<0.001),Fp上清治疗组较结肠炎模型组降低(P<0.01),但仍显著高于正常对照组(P<0.05)(图3)。
四、结肠组织p-NF-κBp65表达
免疫组化染色显示,p-NF-κBp65主要表达于细胞核(图4A-C)。结肠炎模型组结肠组织p-NF-κBp65 免疫组化评分显著高于正常对照组(P<0.001),Fp上清治疗组评分较结肠炎模型组降低(P<0.001),但仍显著高于正常对照组(P<0.05)(图4D)。
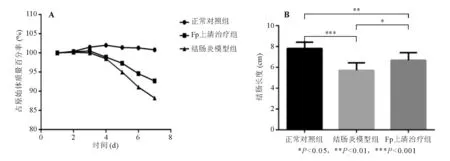
图1 各组小鼠体质量变化和结肠长度比较
*P<0.05,**P<0.01,***P<0.001
五、外周血TNF-α、IL-6、COX-2水平
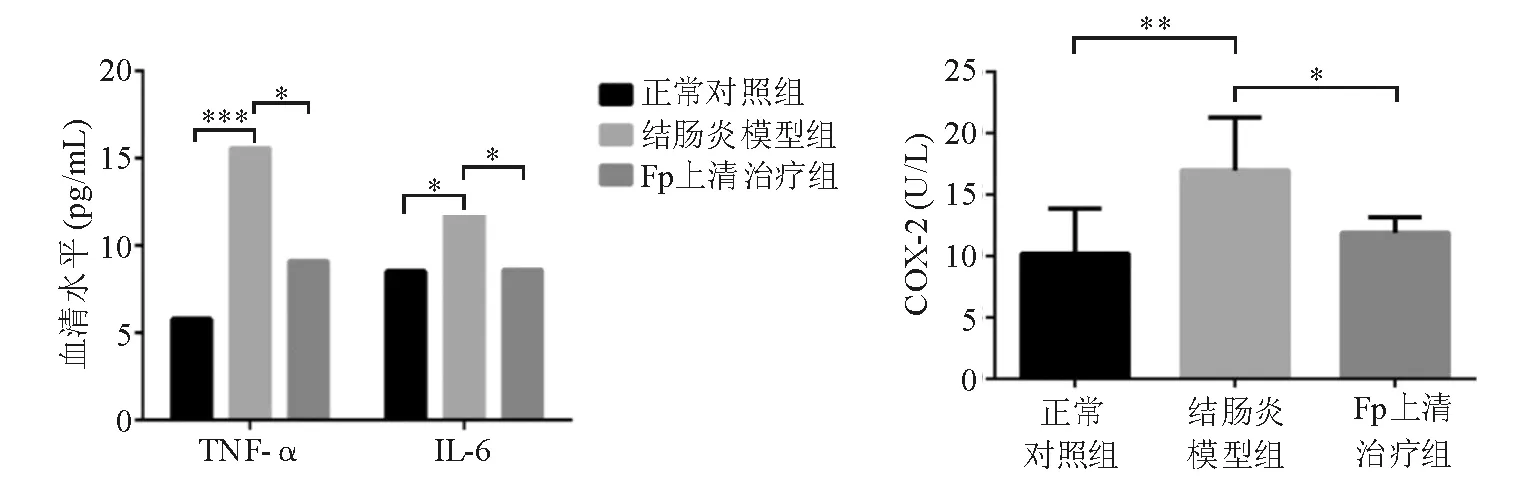
ELISA检测显示,结肠炎模型组血清TNF-α、IL-6、COX-2水平显著高于正常对照组(P<0.05),Fp上清治疗组3种炎症因子的血清水平均较结肠炎模型组显著降低 (P<0.05), 与正常对照组相比差异无统计学意义(P>0.05)(图5)。
讨 论
IBD的病因和发病机制尚未完全明确,但以有益菌减少为特征的肠道微生态失调被认为是IBD的免疫病理学基础[7],益生菌如双歧杆菌、乳酸杆菌常被用于IBD的辅助治疗。本实验结果显示肠道共生菌Fp的上清液能显著减轻小鼠结肠炎模型的结肠炎症,改善结肠缩短、体质量下降等表现,提示其可能成为IBD辅助治疗的又一益生菌类型。

A:正常对照组;B:结肠炎模型组;C:Fp上清治疗组;D:各组结肠组织病理学评分
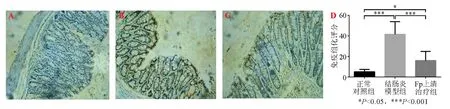
A:正常对照组;B:结肠炎模型组;C:Fp上清治疗组;D:各组p-NF-κB p65免疫组化评分
*P<0.05,**P<0.01,***P<0.001
既往研究[3]发现Fp上清液可阻断NF-κB炎症信号通路激活,进而发挥抗结肠炎作用。转录因子NF-κB广泛存在于真核细胞内,通过调控相关基因转录参与免疫、炎症以及细胞存活、凋亡等重要病理生理过程[8]。人类IBD[9]和小鼠结肠炎模型[10]结肠黏膜中均存在NF-κB过度或不适当地激活,提示其活化可能是IBD发病中的一个重要环节。静息状态下的NF-κB与其抑制蛋白(IκB)结合形成复合物,因其核定位区被覆盖而滞留于胞质内。细胞受脂多糖、细胞因子、免疫刺激剂等外界因素刺激后,IκB被降解,NF-κB发生磷酸化并暴露出核定位区,随即转位至胞核[11],与下游基因的κB位点结合而发挥相应生物学效应。因此本实验通过检测p-NF-κBp65表达强度评估NF-κB的激活程度(p65为NF-κB中具有转录激活功能的亚单位),结果显示予Fp上清液治疗的结肠炎模型小鼠,结肠组织p-NF-κBp65表达强度较未予治疗的模型小鼠显著降低,提示NF-κB活化受抑,同时小鼠临床症状和结肠组织病理学改变明显好转,表明阻断NF-κB激活可有效治疗小鼠结肠炎症。另有研究[12]发现,以NF-κBp65反义寡核苷酸阻断NF-κB激活可预防和治疗小鼠慢性结肠炎相关肠纤维化。结合本研究结果,证实NF-κB可作为IBD治疗的特异性靶点。
TLR4是TLRs家族的重要成员,其可识别肠道微生物产生的细胞毒性物质和内毒素[13],然后通过招募MyD88和TRIF激活NF-κB[5],进而诱导肠上皮细胞分泌免疫炎症因子[13]。TLR4在正常结肠黏膜肠上皮细胞中表达水平极低,在活动期IBD患者肠上皮细胞中则呈强表达[14],伴NF-κB异常激活[9],在本实验和其他动物实验[15-17]中亦观察到类似现象,因此可认为TLR4/NF-κB信号通路与IBD密切相关。乳杆菌L.suntoryeus[16]、柚皮素[17]、黄芩苷[15]在实验性结肠炎中的抗炎效应亦与抑制TLR4/NF-κB信号通路激活有关,与本研究结果相符。
TNF-α、IL-6、COX-2基因增强子或启动子区均含有κB结合位点,其转录受NF-κB调控[18]。本实验结果证实,NF-κB激活后,这些炎症因子的血清水平均显著升高。TNF-α、IL-6是IBD中重要的促炎细胞因子,在活动期IBD[19]和结肠炎动物模型中[15-17]均明显升高,可诱导急性期蛋白产生[20],在黏膜炎症级联反应中起关键作用[21]。抗TNF-α单抗英夫利昔单抗、阿达木单抗临床上用于CD的诱导和维持缓解取得良好疗效。本实验结果显示,结肠炎模型小鼠的血清TNF-α、IL-6水平升高可为Fp上清液治疗所抑制。
COX-2在正常结肠组织中未见表达,在IBD炎症部位的肠上皮细胞和固有层单核细胞[22]以及结肠炎模型动物的肠黏膜和血清中[15,17]则呈高水平表达,提示COX-2参与了结肠炎症的发生、发展。COX-2能催化花生四烯酸生成前列腺素E2(PGE2),而高水平的PGE2可通过调控IL-23/IL-17轴加剧实验性结肠炎的炎症反应过程[23]。尽管多数学者认为COX-2具有促炎作用,但亦存在相悖观点,如有研究发现COX-2基因缺失小鼠对DSS诱导的急性结肠炎敏感性增高[24],而在另一些研究中,抑制COX-2对实验性结肠炎具有保护作用[25]。故有学者提出COX-2在IBD中主要发挥抗炎作用,但此作用主要发生于急性炎症消退期,在此阶段,COX-2可诱导生成具有抗炎作用的PGD2[26-27]。本实验发现Fp上清液治疗可下调结肠炎模型小鼠升高的血清COX-2水平。鉴于COX-2在IBD中作用的复杂性,对涉及COX-2抑制的IBD临床治疗药物,使用前应全面权衡利弊。
既往研究[3-4]发现了一个有趣的现象,即Fp上清液表现出较Fp活菌更明显的抗炎效应,推测发挥抗炎作用的直接效应成分是Fp的某些代谢产物而非活菌本身。本研究直接使用Fp上清液进行实验,发现其在DSS诱导的实验性结肠炎中表现出明显的抗炎效应,此效应与抑制TLR4/NF-κB信号通路激活及其下游炎症因子表达有关。分离并鉴定Fp上清液中发挥抗炎作用的有效成分并将其应用于临床,将是后续研究的重点内容。
1HansenR,RussellRK,ReiffC,etal.MicrobiotaofDe-NovoPediatricIBD:IncreasedFaecalibacterium PrausnitziiandReducedBacterialDiversityinCrohn’sButNotinUlcerativeColitis[J].AmJGastroenterol, 2012, 107 (12): 1913-1922.
2VarelaE,ManichanhC,GallartM,etal.ColonisationbyFaecalibacterium prausnitziiandmaintenanceofclinicalremissioninpatientswithulcerativecolitis[J].AlimentPharmacolTher, 2013, 38 (2): 151-161.
3SokolH,PigneurB,WatterlotL,etal. Faecalibacterium prausnitziiisananti-inflammatorycommensalbacteriumidentifiedbygutmicrobiotaanalysisofCrohndiseasepatients[J].ProcNatlAcadSciUSA, 2008, 105 (43): 16731-16736.
4 洪娜,邱新运,张明明,等. 普拉梭菌对实验性大鼠结肠炎防治的初步研究[J]. 中华消化杂志, 2012, 32 (7):459-465.
5SiddiqueI,KhanI.MechanismofregulationofNa-Hexchangerininflammatoryboweldisease:roleofTLR-4signalingmechanism[J].DigDisSci, 2011, 56 (6): 1656-1662.
6WirtzS,NeufertC,WeigmannB,etal.Chemicallyinducedmousemodelsofintestinalinflammation[J].NatProtoc, 2007, 2 (3): 541-546.
7ComitoD,RomanoC.Dysbiosisinthepathogenesisofpediatricinflammatoryboweldiseases[J].IntJInflam, 2012, 2012: 687143.
8ChakrabortyJB,MannDA.NF-kappaBsignalling:embracingcomplexitytoachievetranslation[J].JHepatol, 2010, 52 (2): 285-291.
9AndresenL,JørgensenVL,PernerA,etal.ActivationofnuclearfactorkappaBincolonicmucosafrompatientswithcollagenousandulcerativecolitis[J].Gut, 2005, 54 (4): 503-509.
10SinghK,ChaturvediR,BarryDP,etal.Theapolipo-proteinE-mimeticpeptideCOG112inhibitsNF-kappaBsignaling,proinflammatorycytokineexpression,anddiseaseactivityinmurinemodelsofcolitis[J].JBiolChem, 2011, 286 (5): 3839-3850.
11DiamantG,DiksteinR.TranscriptionalControlbyNF-κB:ElongationinFocus[J].BiochimBiophysActa, 2013: 937-945.
12LawranceIC,WuF,LeiteAZ,etal.Amurinemodelofchronicinflammation-inducedintestinalfibrosisdown-regulatedbyantisenseNF-kappaB[J].Gastroenterology, 2003, 125 (6): 1750-1761.
13JungHC,EckmannL,YangSK,etal.Adistinctarrayofproinflammatorycytokinesisexpressedinhumancolonepithelialcellsinresponsetobacterialinvasion[J].JClinInvest, 1995, 95 (1): 55-65.
14CarioE,PodolskyDK.Differentialalterationinintestinalepithelialcellexpressionoftoll-likereceptor3 (TLR3)andTLR4ininflammatoryboweldisease[J].InfectImmun, 2000, 68 (12): 7010-7017.
15CuiL,FengL,ZhangZH,etal.Theanti-inflammationeffectofbaicalinonexperimentalcolitisthroughinhibitingTLR4/NF-κBpathwayactivation[J].IntImmuno-pharmacol, 2014, 23 (1): 294-303.
16LeeJH,LeeB,LeeHS,etal. Lactobacillus suntoryeusinhibitspro-inflammatorycytokineexpressionandTLR-4-linkedNF-κBactivationinexperimentalcolitis[J].InternJColorectDis, 2009, 24 (2): 231-237.
17DouW,ZhangJ,SunA,etal.ProtectiveeffectofnaringeninagainstexperimentalcolitisviasuppressionofToll-likereceptor4/NF-κBsignaling[J].BritJNutr, 2013, 110 (4): 599-608.
18HaydenMS,GhoshS.NF-κBinimmunobiology[J].CellRes, 2011, 21 (2): 223-244.
19IshiguroY.Mucosalproinflammatorycytokineproductioncorrelateswithendoscopicactivityofulcerativecolitis[J].JGastroenterol, 1999, 34 (1): 66-74.
20SartorRB.Cytokinesinintestinalinflammation:pathophysiologicalandclinicalconsiderations[J].Gastroenterology, 1994, 106 (2): 533-539.
21BlamME,SteinRB,LichtensteinGR.Integratinganti-tumornecrosisfactortherapyininflammatoryboweldisease:currentandfutureperspectives[J].AmJGastroenterol, 2001, 96 (7): 1977-1997.
22SingerII,KawkaDW,SchloemannS,etal.Cyclooxygenase2isinducedincolonicepithelialcellsininflammatoryboweldisease[J].Gastroenterology, 1998, 115 (2): 297-306.
23SheibanieAF,YenJH,KhayrullinaT,etal.TheproinflammatoryeffectofprostaglandinE2inexperimentalinflammatoryboweldiseaseismediatedthroughtheIL-23 -->IL-17axis[J].JImmunol, 2007, 178 (12): 8138-8147.
24MorteauO,MorhamSG,SellonR,etal.Impairedmucosaldefensetoacutecolonicinjuryinmicelackingcyclooxygenase-1orcyclooxygenase-2[J].JClinInvest, 2000, 105 (4): 469-478.
25DudhgaonkarSP,TandanSK,KumarD,etal.Influenceofsimultaneousinhibitionofcyclooxygenase-2andinduciblenitricoxidesynthaseinexperimentalcolitisinrats[J].Inflammopharmacology, 2007, 15 (5): 188-195.
26ZamunerSR,WarrierN,BuretAG,etal.Cyclooxygenase2mediatespost-inflammatorycolonicsecretoryandbarrierdysfunction[J].Gut, 2003, 52 (12): 1714-1720.
27GilroyDW,Colville-NashPR,WillisD,etal.Induciblecyclooxygenasemayhaveanti-inflammatoryproperties[J].NatMed, 1999, 5 (6): 698-701.
(2016-03-21收稿;2016-04-11修回)
Effect ofFaecalibacteriumprausnitziion TLR4/NF-κB Signaling Pathway in Experimental Colitis
CHENZhaogui1,YUChenggong1,2.
1DepartmentofGastroenterology,theAffiliatedDrumTowerHospitalofNanjingUniversityMedicalSchool,Nanjing(210008);2DepartmentofGastroenterology,YizhengHospital,DrumTowerHospitalGroupofNanjing,Nanjing
Correspondence to: YU Chenggong, Email: chenggong_yu@nju.edu.cn
Background:Faecalibacteriumprausnitzii(Fp) is one of the commensal bacteria colonized in intestine, lowering of Fp is the key character of intestinal microbial dysbiosis in patients with inflammatory bowel disease (IBD). It has been demonstrated that the supernatant of Fp exerts an obvious anti-inflammatory effect in experimental colitis. Aims: To investigate whether the anti-inflammatory effect of Fp supernatant in experimental colitis is related to inhibition of TLR4/NF-κB pathway activation. Methods: Twenty-four C57BL/6 mice were randomly divided into the normal control group, colitis group and Fp supernatant-treated group. Acute colitis was induced by drinking 3.5% dextran sulfate sodium (DSS) in tap water for five days. Weight loss and histopathological damage of colon tissue were observed; real-time PCR and immunohistochemistry were applied to determine the TLR4 mRNA and phosphorylated NF-κB p65 (p-NF-κB p65) expression in colon tissue. Serum levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and cyclooxygenase-2 (COX-2) were detected by ELISA method. Results: Compared with normal control group, mice in colitis group showed a heavier weight loss and shortened colon length; the colonic histopathological score, TLR4 mRNA and p-NF-κB p65 expressions, together with the serum levels of TNF-α, IL-6 and COX-2 were significantly increased (P<0.05). When treated with Fp supernatant, all the above-mentioned indicators were reversed with statistical significance (P<0.05). No significant differences were found in serum inflammatory cytokines between Fp supernatant-treated group and normal control group (P>0.05). Conclusions: The mechanism for Fp supernatant against experimental colitis is related to the inhibition of TLR4/NF-κB pathway activation and suppression of downstream inflammatory cytokines.
Faecalibacteriumprausnitzii; Colitis; Toll-Like Receptor 4; NF-kappa B; Tumor Necrosis Factor-alpha; Interleukin-6; Cyclooxygenase 2
10.3969/j.issn.1008-7125.2016.11.002
国家自然科学基金(81470819)
#Email: a743381985@126.com
&本文通信作者,Email: chenggong_yu@nju.edu.cn
猜你喜欢
现代装饰(2022年5期)2022-10-13
中老年保健(2022年5期)2022-08-24
法律方法(2022年1期)2022-07-21
昆明医科大学学报(2021年4期)2021-07-23
中华养生保健(2020年7期)2020-11-16
中成药(2017年9期)2017-12-19
中成药(2017年10期)2017-11-16
中药与临床(2015年5期)2015-12-17
中国药理学通报(2014年2期)2014-05-09
中医研究(2014年6期)2014-03-11
