
沉默RohA基因对舌鳞状细胞癌细胞增殖的影响
2016-12-21 03:18:01严国鑫樊兵邹荣海张健孙晓峰童磊王奇民韩金宏鲁旭飞王莹周元何宗轩廖奕翔李宁曹蕾陈正岗
华西口腔医学杂志 2016年6期
严国鑫 樊兵 邹荣海 张健 孙晓峰 童磊 王奇民 韩金宏 鲁旭飞 王莹 周元 何宗轩 廖奕翔 李宁 曹蕾 陈正岗,6
1.无锡市第二人民医院口腔科,无锡 214002;
2.青岛大学医学院附属青岛市市立医院口腔医学中心,青岛 266071;
3.即墨市普东卫生院口腔科,青岛 266234;4.潍坊医学院口腔医学院,潍坊 261021;
5.大连医科大学研究生院,大连 116044;
6.上海交通大学医学院附属第九人民医院口腔颌面外科,上海 200011
·肿瘤学专栏·
沉默RohA基因对舌鳞状细胞癌细胞增殖的影响
严国鑫1樊兵1邹荣海1张健1孙晓峰1童磊2王奇民2韩金宏2鲁旭飞3王莹4周元4何宗轩2廖奕翔2李宁5曹蕾5陈正岗2,6
1.无锡市第二人民医院口腔科,无锡 214002;
2.青岛大学医学院附属青岛市市立医院口腔医学中心,青岛 266071;
3.即墨市普东卫生院口腔科,青岛 266234;4.潍坊医学院口腔医学院,潍坊 261021;
5.大连医科大学研究生院,大连 116044;
6.上海交通大学医学院附属第九人民医院口腔颌面外科,上海 200011
目的 通过RNA干扰技术沉默RhoA基因从而探讨RhoA对舌癌细胞增殖和生长的影响及其作用机制。方法 体外培养舌鳞状细胞癌SCC-4细胞,以小分子干扰RNA转染沉默RhoA基因的表达。实验分为3组:实验组(又分为实验1组和实验2组,脂质体分别转染对应序列1的RhoA-siRNA和序列2的RhoA-siRNA)、阴性对照组(脂质体转染NC-siRNA)和空白对照组(不转染siRNA)。采用实时定量聚合酶链反应技术检测SCC-4细胞转染后RhoA mRNA的表达,Western blot检测RhoA、Cyclin D1、p21和p27蛋白的表达,四唑盐比色法检测舌癌细胞生长水平和倍增时间。结果 与阴性对照组和空白对照组相比,实验组舌癌细胞的RhoA基因及蛋白表达降低,p21、p27蛋白表达升高,Cyclin D1蛋白表达降低,细胞倍增时间延长,增殖能力降低(P<0.05)。结论 沉默RhoA基因可以抑制舌癌细胞的增殖和生长,RhoA基因通过调控细胞周期信号转导途径影响舌癌细胞的增殖,RhoA基因可以成为舌癌基因治疗的靶点。
RhoA; 舌癌; 细胞增殖; RNA干扰
口腔癌是头颈部常见的恶性肿瘤,其患病率和病死率占常见恶性肿瘤的第6位[1];舌鳞状细胞癌(以下简称舌癌)约占口腔癌的30%[2]。近几十年来,舌癌患者总的生存率并没有得到显著提高,其5年生存率仍然不足60%,晚期伴有局部扩散或远处转移者预后更差[3]。
Rho蛋白具有三磷酸鸟苷(guanosine triphosphate,GTP)酶活性,在细胞的信号传导通路中作为信号转换器或分子开关,主要通过参与调节肌动蛋白(细胞骨架)的活动、细胞分裂增殖、细胞变性的过程,在细胞黏附、迁移运动和增殖凋亡等多种生物学行为中发挥着重要的作用[4]。已有研究[5-6]证明,RhoA及其下游效应蛋白能够使细胞发生恶性转化、增殖能力增强、抵抗凋亡、易于发生侵袭和转移;其表达的改变和肿瘤的恶性程度密切相关;但这些蛋白通过哪些信号转导途径来发挥其生物学效应,各个效应蛋白的信号网络之间是否存在交互调控等,尚待进一步阐明[7]。针对RhoA干扰其功能,有可能为肿瘤的治疗提供新的策略和途径。本研究应用RNA干扰技术(RNA interference,RNAi)沉默舌癌SCC-4细胞的RhoA基因,观察肿瘤细胞RhoA基因和蛋白水平表达以及肿瘤的增殖活性的变化,并对相关信号通路的蛋白分子进行检测,探讨RhoA基因对肿瘤细胞增殖的影响,为舌癌针对RhoA靶向的基因治疗提供实验基础。
1 材料和方法
1.1 主要试剂及材料
人舌癌细胞株SCC-4由山东大学口腔医学院馈赠。DMEM高糖培养基、乙二胺四乙酸(ethylenedi aminetetraacetic acid,EDTA)/胰蛋白酶、Trizol、阳离子脂质体试剂Lipofectamine2000(Invitrogen公司,美国),胎牛血清、青霉素/链霉素(Gibco公司,美国),SYBR Premix Ex TaqTM(Perfect Real Time)试剂盒(Takara公司,日本),RhoA鼠抗人单克隆抗体(Santa Cruz公司,美国),辣根过氧化物酶(horseradish peroxidase,HRP)标记的羊抗鼠二抗(北京中杉金桥生物技术有限公司),放射免疫沉淀法(radio immunoprecipitation assay,RIPA)蛋白裂解液(江苏碧云天生物技术研究所)。小干扰RNA(small interfering RNA,siRNA)序列合成及基因测序购自上海博尚生物技术有限公司。
1.2 舌癌细胞培养及传代
SCC-4细胞用含10%胎牛血清、青霉素100 mg·mL-1和链霉素100 mg·mL-1的DMEM培养基在37 ℃、5% CO2培养箱培养,向培养瓶(25 cm2)内加入2 mL胰蛋白酶。消化在37 ℃环境下进行,消化2~5 min后把培养瓶放置显微镜下观察,发现胞质回缩、细胞间隙增大后,立即加入5 mL含有血清的DMEM培养基终止消化。用弯头吸管吸取瓶内培养液,反复吹打瓶壁细胞,吹打过程按顺序进行,确保所有底部均被吹到。动作轻柔,尽可能避免出现泡沫。细胞脱离瓶壁后形成细胞悬液。计数,按1∶1比例传代接种在新的培养瓶中。
1.3 RhoA siRNA的合成
登录Genebank数据库确定人RhoA基因序列,序列号为NM_001664,针对RhoA的基因序列设计2条RhoA-siRNA和1条阴性对照序列(NC-siRNA)(表1)。
1.4 siRNA转染沉默RhoA基因
转染前1 d按每孔1×105个分别将舌癌SCC-4细胞接种于不同的24孔板中,每孔培养基500 μL,不含抗生素。贴壁细胞融合率达30%~50%时进行转染。用50 μL Opti-MEI低血清(或无血清)培养基稀释20 pmol siRNA(转染时siRNA终浓度为33 nmol·L-1),轻轻混匀;使用前轻轻摇匀Lipofectamine2000,然后取1 μL Lipofectamine2000在50 μL Opti-MEI培养基中稀释,室温孵育5 min。将前两步所稀释的siRNA和Lipofectamine2000混合(使总体积为100 μL),轻轻混匀,室温放置20 min。每孔细胞中加入100 μL转染液,轻轻摇匀。37 ℃培养48 h后检测基因表达。实验分为3组,实验组、阴性对照组、空白对照组,其中实验组为转染RhoA-siRNA组,脂质体转染RhoA-siRNA,又分为实验1组和实验2组,分别对应序列1的RhoA-siRNA和序列2的RhoA-siRNA;阴性对照组为转染NC-siRNA组,脂质体转染NC-siRNA;空白对照组为转染脂质体Lipo组,仅加入转染混合液,不转染任何的siRNA。

表1 针对RhoA基因序列设计的2条RhoA-siRNA和1条阴性对照序列Tab 1 Two RhoA-siRNA sequences and one negative control sequence designed for RhoA gene
1.5 实时荧光定量聚合酶链反应(quantitative real- time polymerase chain reaction,qRT-PCR)技术检测各组RhoA mRNA的表达
按照Trizol试剂盒提取细胞总RNA,紫外分光光度计测定RNA含量,取5 μL总RNA,在M-MLV反转录酶作用下合成cDNA,再取5 μL反转录产物进行PCR扩增反应,以磷酸甘油醛脱氢酶(glyceraldehyde phosphate dehydrogenase,GAPDH)为内参照。RhoA基因的引物序列,上游:5’-TTCCATCGACAGCCCTGATAGTTTA-3’,下游:5’-CACGTTGGGACAGAAATGCTTG-3’;GAPDH基因的引物序列,上游:5’-GCACCGTCAAGGCTGAGAAC-3’,下游:5’-TGGTGAAGACGCCAGTGGA-3’。PCR反应条件:95 ℃ 30 s;然后95 ℃ 5 s,60 ℃ 34 s,40个循环。荧光信号实时监测和数据分析由Stratagene qRTPCR仪自动完成,采用2-∆∆Ct公式计算RhoA mRNA的相对表达水平,其中Ct值为循环阈值。
1.6 蛋白质免疫印迹法(Western blot)检测目的蛋白的表达
弃培养液后PBS冲洗细胞3次,用RIPA蛋白裂解液裂解,操作于冰上进行,4 ℃下10 000 r·min-1(离心半径为4 cm)离心5 min取上清液,聚氰基丙烯酸正丁酯(bicinchoninic acid,BCA)法测蛋白浓度后,100 ℃变性10 min。用12%聚丙烯酰胺凝胶分离,再电转至硝酸纤维素膜上,5%脱脂奶粉封闭2 h后,单克隆抗体RhoA(1∶200)、细胞周期素D1(Cyclin D1)(1∶1 000)、细胞周期蛋白依赖型激酶抑制因子p21(1∶200)、p27(1∶200)、β-actin(1∶1 000)4 ℃孵育过夜,三羟甲基氨基甲烷缓冲盐水(triethanolamine buffered saline,TBS-T)洗涤,辣根过氧化物酶(horseradish peroxidase,HRP)标记的山羊抗小鼠二抗(1∶5 000)室温孵育2 h,PBS洗涤,加入电化学发光(electrochemiluminescence,ECL)发光液显色曝光。
1.7 四唑盐比色法(methyl thiazolyl tetrazolium,MTT)检测舌癌细胞生长水平和倍增时间的计算
取对数生长期的细胞,分别以每孔1×103个的密度接种于96孔板,设5个复孔,转染RhoA-siRNA后分别连续培养1、2、3 d,每孔加入MTT液(5 g·L-1)20 μL,37 ℃孵育4 h,吸出上清液,加入150 μL二甲基亚砜(dimethyl sulfoxide,DMSO),充分振荡10 min后于酶联免疫检测仪上490 nm比色测吸光度值,以时间为横轴、吸光度值为纵轴,绘制细胞生长曲线。群体倍增时间按Patterson公式计算[8]:Td= T×lg2/lg(NT/N0),Td为倍增时间(h),T为细胞数由N0增至NT所用的时间,N0为接种时的细胞数,NT为培养T小时后的细胞数。
1.8 统计学分析
采用SPSS 12.0软件进行统计分析,计量资料的组间比较用t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 舌癌细胞RhoA基因的表达
实时荧光定量PCR检测结果(图1)显示,与阴性对照组、空白对照组相比,实验组RhoA mRNA表达降低(P<0.05)。
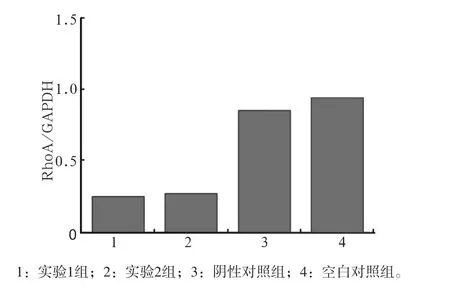
图1 siRNA转染沉默RhoA基因48 h后RhoA mRNA的表达Fig 1 Expression of RhoA mRNA 48 h after RhoA-siRNA transfection
2.2 舌癌细胞RhoA、Cyclin D1、p21和p27蛋白的表达
Western blot检测结果显示,与阴性对照组、空白对照组相比,实验组的RhoA、Cyclin D1蛋白的表达降低,p21、p27蛋白的表达升高(图2)(P<0.05)。
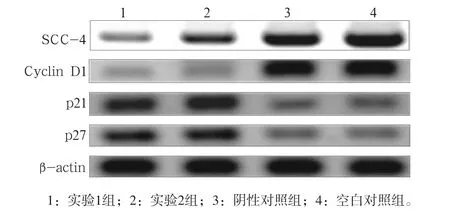
图2 siRNA转染沉默RhoA基因48 h后RhoA、Cyclin D1、p21和 p27蛋白的表达 Fig 2 Expression of RhoA, Cyclin D1, p21 and p27 protein 48 h after RhoA-siRNA transfection
2.3 舌癌细胞生长水平
MTT法对细胞活力检测结果表明,与阴性对照组和空白对照组相比,实验组SCC-4细胞的增殖能力降低(图3);实验组的倍增时间(实验1组29.7 h± 1.7 h,实验2组228.4 h±3.5 h)较对照组(阴性对照组20.3 h±3.6 h,空白对照组21.2 h±2.8 h)延长(P<0.05)。
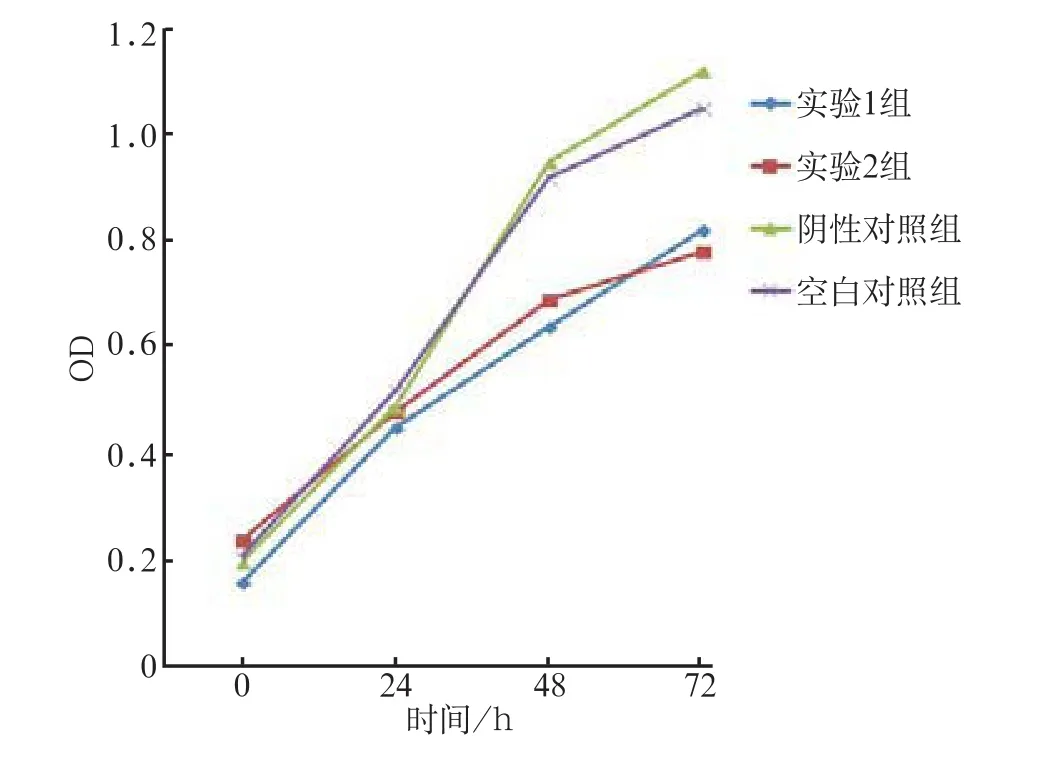
图3 siRNA转染沉默RhoA基因各组不同时间细胞的增殖活力 Fig 3 MTT assay shows the changes of the cell proliferation ability after RhoA silencing
3 讨论
Rho亚家族是一组相对分子质量为20×103~30×103的鸟苷酸结合蛋白,具有GTP酶活性。Rho家族蛋白在非活性GDP结合形式和活性GTP结合形式之间循环,具有RhoA、RhoB和RhoC三种异构体。RhoA、RhoB和RhoC高度同源,约有85%的氨基酸序列相同[9]。RhoA在多种肿瘤组织及细胞系中如结肠癌、乳腺癌、肺癌、胰腺癌、头颈癌等高表达,在口腔鳞癌中的高表达也已经得到证实[10],推测RhoA与舌癌的发生发展关系密切。
本研究结果显示,RhoA基因沉默后,舌癌细胞的活力明显下降,增殖受到抑制。同时检测与细胞增殖的相关蛋白表达,发现Cyclin D1蛋白表达显著降低,而p21和p27表达显著上升,提示RhoA基因可能通过细胞周期的调控而影响肿瘤的生长。
细胞的增殖周期是一个连续的发展过程,参与细胞周期调控的主要分子有:细胞周期蛋白(Cyclins)、细胞周期蛋白依赖性激酶(Cyclin-dependent kinases,CDKs)和周期蛋白依赖性激酶抑制物(Cyclin-dependent kinase inhibitor protein,CDKI)。Cyclins对细胞周期具有正调控作用。现已发现,在Cyclin家族中,Cyclin D1在细胞增殖过程中的意义最大,是G1期细胞增殖信号的关键蛋白[11]。Cyclin D1是最早发现的原癌基因[12],其表达与多种肿瘤的发生密切相关[13],在口腔癌的研究中,也证实了Cyclin D1在口腔癌中高表达,与肿瘤的发生、发展、临床分期、病理分级、淋巴结转移以及预后关系密切,可以成为口腔癌预后判断及治疗方案选择的重要生物标志物[14-15]。Cyclin D1在G1期的正性调控作用是通过Cyclin D1与CDK4/CDK6结合并使之激活,导致视网膜母细胞瘤蛋白(retinoblastoma protein,pRb)在G1-S期发生磷酸化,从而使核转录因子从pRb抑制下解离,进一步激活这些基因的表达,启动DNA复制,推动细胞从G1期进入S期[16]。Cyclin D1的基因扩增和过表达可促进G1/S期转化,加速细胞周期,使细胞持续增殖导致恶性转化。本研究中RhoA基因沉默后,Cyclin D1的表达显著下降,抑制了Cyclin D1在G1期的表达,细胞将不能进入S期,细胞的增殖和肿瘤的生长则会受到明显的抑制。
作为抑癌基因,CDK抑制因子p21和p27是两个重要的细胞周期调控因子。p21在正常细胞DNA受损伤时,被p53诱导产生,随之抑制DNA复制,增强DNA修复,确保遗传物质精确地传递给下一代,以消除由于DNA损伤的积累而引起肿瘤发生的隐患[17]。p21的表达减弱或消失可能使抑制细胞增殖的作用减弱,或细胞正常增生转变为增生分化不良,导致肿瘤发生的可能[18]。p27作为细胞中G1-S期限制点的重要负性调控因子,通过与Cyclin竞争,或直接与CDK或Cyclin-CDK复合物结合,抑制CDK催化活性,阻止细胞周期G1/S期转换,抑制细胞增生。当p27基因表达异常时,可导致细胞无限增殖和肿瘤的形成;p27水平的降低会促进G1期的进展,增加细胞增殖的速度。许多研究[19-21]表明,在人类几乎所有的上皮速度。许多研究[19-21]表明,在人类几乎所有的上皮性肿瘤中都有p27蛋白表达的下降或缺失,p27表达降低已经在多种肿瘤中观察到并得到证实。本研究中,通过抑制RhoA的基因表达使p27过度表达,抑制了组蛋白H1的磷酸化,从而抑制了CDK的活性,抑制了细胞进入细胞周期的S期。从功能上讲,p21和p27是调控细胞周期的相关基因,同属于CDKI,其在氨基酸水平上有着明显的同源性。本研究证实,RhoA基因通过调控Cyclin D1、p21和p27来实现对细胞周期的调控。
RhoA可以促进Cyclin D1的转录。Cyclin D1表达水平受多种信号通路调控,目前研究比较详细的是RAS-RAF-MEK-p42/p44 MAPK信号通路。p42/p44 MAPK信号通路的持续激活可以使Cyclin D1高表达,而高水平的Cyclin D1对于维持肿瘤的功能十分重要;整联蛋白(Integrin)介导的黏着斑复合物的形成需要与细胞骨架相连,因此黏着斑复合物的形成依赖于RhoA蛋白的活性。研究[22]同样证实,Integrin的聚集和黏着斑复合物的形成对于G1/S进程相当重要。例如在NIH3T3成纤维细胞中,RhoA可以促进黏着斑的形成,同时也可以被Integrin激活,进而激活下游的效应分子ROCK激酶,ROCK磷酸化LIM激酶,这一连锁信号在G1期促进持续的p42/p44 MAPK激活和Cyclin D1的表达[23],因此,RhoA在G1期对于Cyclin D1的适时表达起到了至关重要的调节作用。此外,RhoA对细胞周期的调控作用还可以通过调控p21和p27蛋白来实现。研究发现,过表达激活的RhoA可以促进促有丝分裂原刺激下的G1期细胞中p27的降解[23];通过使用抑制素、细菌毒素和过表达显性失活的RhoA,都可使p27在细胞中聚集,使细胞周期进程停滞[24]。p27蛋白的降解通常是通过CDK2在G1后期介导的磷酸化引起的,所以RhoA蛋白可能并不是直接与p27蛋白发生相互作用,而是通过影响G1蛋白例如Cyclin D1的表达,进而激活Cyclin E/CDK2,最终导致p27蛋白降解[25];抑制RhoA的活性还可以增加p27基因的转录[26]。通过使用Rho蛋白异戊烯化抑制剂GGTI不仅可以阻碍G1/S进程而且还伴有p21蛋白的表达上调,但是RhoA信号对于p21蛋白表达调控的精确机制还有待于进一步研究[27]。RhoA可能在转录水平抑制p21蛋白表达,也可能是通过Ras介导p42/p44 MAPK信号途径的激活[28]。
综上所述,RhoA蛋白在舌癌的发生发展中具有重要的作用,可以参与细胞周期的调控,通过调节Cyclin D1、p21和p27来促进肿瘤细胞的增殖。RhoA蛋白有可能成为舌癌治疗的新的靶点。但本研究尚存在一定的不足,本研究仅从体外实验的角度探讨了RhoA基因调控细胞周期的可能机制,尚需体内实验来进一步加以阐明和证实。
[1] Humphrey PA, Wong AJ, Vogelstein B, et al. Amplification and expression of the epidermal growth factor receptor gene in human glioma xenografts[J]. Cancer Res, 1988, 48 (8):2231-2238.
[2] Ferrari D, Codecà C, Fiore J, et al. Biomolecular markers in cancer of the tongue[J]. J Oncol, 2009, 2009:412908.
[3] Bodner L, Manor E, Friger MD, et al. Oral squamous cell carcinoma in patients twenty years of age or younger—review and analysis of 186 reported cases[J]. Oral Oncol, 2014, 50 (2):84-89.
[4] Kutys ML, Yamada KM. An extracellular-matrix-specific GEF-GAP interaction regulates Rho GTPase crosstalk for 3D collagen migration[J]. Nat Cell Biol, 2014, 16(9):909-917.
[5] Fujita M, Imadome K, Endo S, et al. Nitric oxide increases the invasion of pancreatic cancer cells via activation of the PI3K-AKT and RhoA pathways after carbon ion irradiation [J]. FEBS Lett, 2014, 588(17):3240-3250.
[6] Hwang H, Kim EK, Park J, et al. RhoA and Rac1 play independent roles in lysophosphatidic acid-induced ovarian cancer chemotaxis[J]. Integr Biol (Camb), 2014, 6(3):267-276.
[7] Wacker I, Behrens J. Activin B antagonizes RhoA signaling to stimulate mesenchymal morphology and invasiveness of clear cell renal cell carcinomas[J]. PLoS ONE, 2014, 9(10): e111276.
[8] 钱静, 陈不尤, 刘贤称, 等. 人肺腺癌厄洛替尼耐药细胞系PC-9/ER的建立及其特性[J]. 临床与病理杂志, 2015, 35(6):1080-1086.
Qian J, Chen BY, Liu XC, et al. Establishment and characterization of a erlotinib-drug resistant variant of human lung adenocarcinoma cell line PC-9/ER[J]. Int J Pathol Clin Med, 2015, 35(6):1080-1086.
[9] Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration[J]. J Microsc, 2013, 251(3):242-249.
[10] 周峻, 何勇, 董绍忠, 等. RhoA小干扰RNA对舌癌细胞Tca8113增殖、侵袭影响的体外实验[J]. 中华口腔医学杂志, 2010, 45(9):520-524.
Zhou J, He Y, Dong SZ, et al. Effects of rhoa sirna on the proliferation, adhesion, migration and invasion of tongue squamous cell carcinoma tca8113 cells in vitro[J]. Chin J Stomatol, 2010, 45(9):520-524.
[11] Casimiro MC, Velasco-Velázquez M, Aguirre-Alvarado C, et al. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present[J]. Expert Opin Investig Drugs, 2014, 23(3):295-304.
[12] Bartkova J, Lukas J, Strauss M, et al. Cell cycle-related variation and tissue-restricted expression of human cyclin D1 protein[J]. J Pathol, 1994, 172(3):237-245.
[13] Pestell RG. New roles of cyclin D1[J]. Am J Pathol, 2013, 183(1):3-9.
[14] Zhao Y, Yu D, Li H, et al. Cyclin D1 overexpression is associated with poor clinicopathological outcome and survival in oral squamous cell carcinoma in Asian populations: insights from a meta-analysis[J]. PLoS ONE, 2014, 9(3): e93210.
[15] Ramakrishna A, Shreedhar B, Narayan T, et al. Cyclin D1 an early biomarker in oral carcinogenesis[J]. J Oral Maxillofac Pathol, 2013, 17(3):351-357.
[16] Jirawatnotai S, Hu Y, Livingston DM, et al. Proteomic identification of a direct role for cyclin d1 in DNA damage repair [J]. Cancer Res, 2012, 72(17):4289-4293.
[17] Yousefi B, Rahmati M, Ahmadi Y. The roles of p53R2 in cancer progression based on the new function of mutant p53 and cytoplasmic p21[J]. Life Sci, 2014, 99(1/2):14-17.
[18] Ohashi K, Nemoto T, Eishi Y, et al. Expression of the cyclin dependent kinase inhibitor p21WAF1/CIP1 in oesophageal squamous cell carcinomas[J]. Virchows Arch, 1997, 430(5): 389-395.
[19] Bochis OV, Irimie A, Pichler M, et al. The role of Skp2 and its substrate CDKN1B (p27) in colorectal cancer[J]. J Gastrointestin Liver Dis, 2015, 24(2):225-234.
[20] Okutur K, Bassulu N, Dalar L, et al. Predictive and prognostic significance of p27, Akt, PTEN and PI3K expression in HER2-positive metastatic breast cancer[J]. Asian Pac J Cancer Prev, 2015, 16(7):2645-2651.
[21] Ouyang Y, Gao P, Zhu B, et al. Downregulation of micro-RNA-429 inhibits cell proliferation by targeting p27Kip1 in human prostate cancer cells[J]. Mol Med Rep, 2015, 11 (2):1435-1441.
[22] Zscheppang K, Kurth I, Wachtel N, et al. Efficacy of beta1 integrin and EGFR targeting in sphere-forming human head and neck cancer cells[J]. J Cancer, 2016, 7(6):736-745.
[23] Croft DR, Olson MF. The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms[J]. Mol Cell Biol, 2006, 26(12):4612-4627.
[24] Zhong WB, Hsu SP, Ho PY, et al. Lovastatin inhibits proliferation of anaplastic thyroid cancer cells through upregulation of p27 by interfering with the Rho/ROCK-mediated pathway[J]. Biochem Pharmacol, 2011, 82(11):1663-1672.
[25] Hu W, Bellone CJ, Baldassare JJ. RhoA stimulates p27(Kip) degradation through its regulation of cyclin E/CDK2 activity[J]. J Biol Chem, 1999, 274(6):3396-3401.
[26] Hsu YH, Chang CC, Yang NJ, et al. RhoA-mediated inhibition of vascular endothelial cell mobility: positive feedback through reduced cytosolic p21 and p27[J]. J Cell Physiol, 2014, 229(10):1455-1465.
[27] Hamada M, Miki T, Iwai S, et al. Involvement of RhoA and RalB in geranylgeranyltransferase I inhibitor-mediated inhibition of proliferation and migration of human oral squamous cell carcinoma cells[J]. Cancer Chemother Pharmacol, 2011, 68(3):559-569.
[28] Olson MF, Paterson HF, Marshall CJ. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/ Cip1[J]. Nature, 1998, 394(6690):295-299.
(本文编辑 李彩)
Effects of RhoA silencing on proliferation of tongue squamous cancer cells
Yan Guoxin1, Fan Bing1, Zou Ronghai1, Zhang Jian1, Sun Xiaofeng1, Tong Lei2, Wang Qimin2, Han Jinhong2, Lu Xufei3, Wang Ying4, Zhou Yuan4, He Zongxuan2, Liao Yixiang2, Li Ning5, Cao Lei5, Chen Zhenggang2,6. (1. Dept. of Stomatology, Wuxi No 2. People’s Hospital, Wuxi 214002, China; 2. Center of Stomatology, Qingdao Municipal Hospital Affiliated to Qingdao University Medical College, Qingdao 266071, China; 3. Dept. of Stomatology, Pudong Healthcare Center of Jimo County, Qingdao 266234, China; 4. College of Stomatology, Weifang Medical University, Weifang 261021, China; 5. Postgraduate School, Dalian Medical University, Dalian 116044, China; 6. Dept. of Oral and Maxillofacial Surgery, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200011, China)
Supported by: The National Natural Science Foundation of China (81372908); Major Project of Science and Technology Grant of Nanjing Medical University (2012NJMU248); Project of Qingdao Municipal Health and Family Planning Commission (2014-WJZD009, 2013-WSZD011). Correspondence: Chen Zhenggang, E-mail: chenzhg1973@163.com.
Objective This study investigated the effect of RhoA silencing through RNA interference on proliferation and growth of tongue cancer cells, as well as explored the possible mechanisms of this effect. Methods SSC-4 tongue cancer cells were cultured in vitro and then transfected with small interfering RNA to knock down RhoA expression. The tested cells were divided into three groups: experimental group (experimental group 1: transfected with RhoA-siRNA-1; experimental group 2: transfected with RhoA-siRNA-2), negative control group (transfected by random sequence NC-siRNA), and blank control group (transfected with Lipofectamine).The expression levels of RhoA mRNA were respectively measured by quantitative real-time polymerase chain reaction and western blot assay. Moreover, the expression levels of cyclin D1, p21, and p27 and RhoA protein were evaluated by Western blot assay. Proliferation and growth potentiality were analyzed through evaluation of doubling times and methyl thiazolyl tetra-zolium assessment. Results The expression levels of RhoA gene and protein of experimental groups significantly decreased following siRNA transfection compared with those in the negative and blank control groups. The expression of cyclin D1 decreased significantly and that of p21 and p27 increased significantly. The doubling time was extended and the growth potentiality decreased. Conclusion The results indicated that RhoA silencing can inhibit proliferation of tongue cancer cells, whereas RhoA affects cell proliferation by regulating the cell cycle pathway. Thus, RhoA is a potential target in gene therapy for tongue cancer.
RhoA; tongue cancer; cell proliferation; RNA interference
R 739.86
A
10.7518/hxkq.2016.06.014
2016-02-16;
2016-06-10
国家自然科学基金(81372908);南京医科大学科技发展基金重点项目(2012NJMU248);青岛市卫计委计划项目(2014-WJZD009,2013-WSZD011)
严国鑫,副主任医师,硕士,E-mail: yanguo_xin2015@ 163.com
陈正岗,副主任医师,博士,E-mail: chenzhg1973@163. com
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17 07:30:02
中华养生保健(2020年1期)2020-11-16 00:47:56
奥秘(创新大赛)(2019年9期)2019-10-09 02:03:56
小哥白尼(趣味科学)(2019年1期)2019-04-12 00:23:56
国际呼吸杂志(2019年4期)2019-03-12 01:07:30
实用口腔医学杂志(2017年6期)2017-09-19 02:51:06
奥秘(2017年5期)2017-07-05 11:09:30
家庭用药(2016年7期)2016-05-14 09:39:16
中华老年多器官疾病杂志(2016年7期)2016-04-28 08:43:05
癌症进展(2016年10期)2016-03-20 13:15:43
