
曲古抑菌素A对博莱霉素致肺纤维化小鼠NF-κB表达的影响研究
2016-12-21 08:03黄旭晴徐长青李旭
浙江医学 2016年16期
黄旭晴 徐长青 李旭
曲古抑菌素A对博莱霉素致肺纤维化小鼠NF-κB表达的影响研究
黄旭晴 徐长青 李旭
目的 观察曲古抑菌素A(TSA)对博莱霉素(BLM)诱导的肺纤维化小鼠核因子-κB(NF-κB)表达的影响。方法96只健康雄性C57BL/6小鼠随机分为对照组、模型组、TSA早期干预组和TSA晚期干预组,各24只。模型组、TSA早期干预组和TSA晚期干预组小鼠均予以气管内滴注BLM(10mg/kg体重)溶液0.15m l诱导肺纤维化模型,对照组小鼠予0.9%氯化钠注射液0.15m l气管内滴注。建模后,TSA干预组分阶段(早期:第1~14天;晚期:第15~28天)予以TSA(40mg/kg体重)溶液腹腔注射,2次/周。模型组和对照组予以等体积二甲基亚砜腹腔注射作为溶媒对照。建模后第7、14、28天各组均随机分3批处死小鼠8只,留取右肺上叶组织行HE染色和Masson染色观察肺泡炎及肺纤维化程度,并行Szapiel评分,右肺中、下叶组织碱水解法检测羟脯氨酸(HYP)含量,免疫组化方法检测NF-κB表达水平。结果 病理学检查比较,对照组小鼠肺泡结构正常,模型组小鼠肺组织在第7天出现出血、渗出等肺泡炎性改变,第28天出现明显肺纤维化。模型组小鼠肺组织各时点肺泡炎Szapiel评分、HYP含量、NF-κB表达水平均高于对照组(均P<0.05);TSA早期干预组小鼠各时点以上指标均低于模型组(均P<0.05),而TSA晚期干预组与模型组比较均无统计学差异(均P>0.05)。结论 早期应用TSA或能通过抑制NF-κB的表达发挥延缓肺纤维化的作用。
曲古抑菌素A 肺间质纤维化 核因子-κB
特发性肺纤维化是一种病因不明、以进展性的肺纤维化炎症反应为特征的肺间质疾病,其发病率约为0.1‰,且呈逐年增长趋势[1]。特发性肺纤维化可见于各年龄段患者,预后不良,中位生存时间仅有3年[2]。研究显示,肺泡上皮细胞的过度凋亡参与了肺纤维化的发生[3],而转化生长因子β(TGF-β)通过激活其下游效应分子Smad2或Smad3在肺纤维化中也发挥重要作用[4]。核因子-κB(NF-κB)可激活TGF-β,促进成纤维细胞Ⅰ、Ⅲ型胶原蛋白的表达,介导肺泡炎和肺纤维化;而血管紧张素转化酶抑制剂(ACEI)可下调NF-κB的表达,抑制器官纤维化[5]。曲古抑菌素A(TSA)为链霉菌代谢产物,是一种组蛋白脱乙酰基转移酶抑制剂。TSA已被报道可以抑制低氧或血管内皮生长因子诱导的新生血管形成[6],但其对组织纤维化影响的研究还未深入。基于此,本研究以博莱霉素(BLM)致肺纤维化小鼠模型[7]为实验对象,探讨TSA对BLM诱导的肺纤维化小鼠NF-κB表达的影响,以期为临床防治肺纤维化提供参考,现报道如下。
1 材料和方法
1.1 材料 实验动物:健康SPF级、雄性C57BL/6小鼠(南京君科生物工程有限公司)96只,6~8周龄,体重18~22g。主要试剂:TSA(美国Sigma公司),BLM(日本化药株式会社),NF-κB p65试剂盒(武汉博士德生物有限公司),羟脯氨酸(HYP)检测试剂盒(南京建成生物研究所),Masson染液试剂盒(武汉博士德公司)。
1.2 方法
1.2.1 动物分组 小鼠饲养于SPF级动物房,温度23~27℃,湿度55%~65%,12h光照/黑暗交替;适应性饲养1周后,按随机数字表法将小鼠分为4组(模型组、TSA早期干预组、TSA晚期干预组、对照组),每组各24只。
1.2.2 肺纤维化小鼠模型建立和干预 肺纤维化小鼠模型建立:4组小鼠均予颈正中切开,钝性分离浅筋膜逐层暴露气管;模型组、TSA早期干预组、TSA晚期干预组小鼠均予一次性快速气管内滴注BLM(10mg/kg体重)溶液0.15ml,对照组小鼠予一次性快速气管内滴注0.9%氯化钠注射液0.15ml;而后将4组小鼠均直立、快速旋转2min,使药液尽可能均匀分布于肺组织。建模后,TSA早期干预组小鼠于第1~14天予TSA(40mg/kg体重)溶液腹腔注射,2次/周;TSA晚期干预组小鼠于第15~28天予TSA(40mg/kg体重)溶液腹腔注射,2次/周;模型组和对照组小鼠于同时间点予等体积二甲基亚枫(DMSO)腹腔注射作为溶媒对照。
1.3 观察指标 4组小鼠均分别于建模后第7、14、28天随机处死8只。小鼠予氯胺酮腹腔注射麻醉后开胸,分离气管和肺组织;迅速将右肺上叶组织置于4%多聚甲醛中固定,制备石蜡切片,一部分作HE染色及Masson染色进行病理学观察,另一部分作免疫组化染色检测NF-κB表达水平;留取右肺中、下叶组织,剪碎后制成匀浆于-20℃冰箱保存,用于检测肺组织HYP含量。
1.3.1 肺组织病理学观察 肉眼观察小鼠肺组织大体病理学表现,光镜下HE染色观察肺泡炎和肺纤维化程度,以Szapiel评分方法判定肺泡炎和肺纤维化程度[8]。肺泡炎分级:0级示正常肺泡形态,计1分;Ⅰ级示轻度肺泡炎,计2分;Ⅱ级示中度肺泡炎,计3分;Ⅲ级示重度肺泡炎,计4分。肺纤维化分级:0级示正常肺组织,无纤维化;Ⅰ级示轻度纤维化,受累面积<20%,纤维化累及胸膜、胸膜下肺间质,肺泡结构发生紊乱;Ⅱ级示中度纤维化,病变范围约占全肺20%~50%,肺纤维化区域从胸膜开始延伸,但仍属局部性;Ⅲ级示重度纤维化,病变范围>50%,并有融合,可见大小不等的囊气腔,并伴有广泛肺实质结构紊乱。光镜下Masson染色观察肺组织胶原沉积情况,通过测定组织积分光密度(IOD)值进行半定量分析。
1.3.2 肺组织HYP含量检测 取冻存的肺组织(湿质量30~40mg/只)剪碎后制成匀浆,采用碱水解法检测组织HYP含量(mg/g肺组织),具体操作严格按HYP检测试剂盒说明书进行。
1.3.3 肺组织NF-κB表达水平检测 采用免疫组化SP法染色,肺组织切片皆采用图像分析软件先进行颜色分离,然后自动计算其IOD值进行半定量分析。
1.4 统计学处理 应用SPSS16.0统计软件;计量资料以 表示,多组间比较采用方差分析,两两比较采用LSD-t检验。
2 结果
2.1 4组小鼠各时点肺组织大体病理学表现比较 对照组小鼠各时点肺组织均湿润、有弹性、表面光滑,呈粉红色。模型组小鼠第7天时肺组织呈暗红色,可见较多点状病灶;第14天时肺组织出现较多片状白色病灶,肺体积缩小,肺弹性下降;第28天时肺组织呈苍白色,肺体积明显缩小,弹性明显降低,硬度增加,可见散在的细小出血点,表面不光滑,凹凸不平。TSA早期干预组小鼠第28天时肺部湿润、有弹性、表面光滑,体积缩小不明显,弹性尚好,表面轻微结节样改变。TSA晚期干预组小鼠第28天时病变与模型组小鼠相似。
2.2 4组小鼠各时点肺组织病理学表现比较
2.2.1 HE染色病理学表现 对照组小鼠各时点均肺泡壁完整,肺泡腔内无明显渗出。模型组小鼠出现肺泡、肺间质及支气管壁不同程度的纤维增厚,肺部结构明显破坏的病理改变;第7天肺泡中可见到巨噬细胞、单核细胞等炎性细胞浸润;第14天时仍可见少数炎性细胞浸润,部分肺泡萎缩、闭合,成纤维细胞增加;第28天肺泡炎减轻,呈现弥漫性纤维化改变,少见炎性细胞浸润。与模型组比较,TSA早期干预组小鼠第7、14、28天时肺泡炎和肺纤维化均不明显;TSA晚期干预组小鼠与模型组比较,表现相似,第7、14天时仍出现明显巨噬细胞和淋巴细胞等炎性细胞浸润和纤维化改变,第28天时肺组织结构破坏明显、炎性细胞浸润减少,而肺间质大量胶原沉积。4组小鼠各时点肺泡炎Szapiel评分比较见表1。
由表1可见,4组小鼠各时点肺泡炎Szapiel评分比较均有统计学差异(均P<0.05)。与对照组比较,TSA早期干预组、TSA晚期干预组、模型组小鼠各时点肺泡炎Szapiel评分均升高(均P<0.05)。与模型组比较,TSA早期干预组小鼠各时点肺泡炎Szapiel评分均降低(均P<0.05),TSA晚期干预组小鼠各时点肺泡炎Szapiel评分差异均无统计学意义(均P>0.05)。与TSA晚期干预组比较,TSA早期干预组小鼠各时点肺泡炎Szapiel评分亦均降低(均P<0.05)。
2.2.2 Masson染色病理学表现 对照组小鼠肺组织各时点均未见蓝色胶原沉积。模型组小鼠肺组织各时点均可见蓝色胶原沉积,并随着小鼠生存时间延长,蓝色胶原沉积加重。TSA早期干预组小鼠肺组织各时点也可见蓝色胶原沉积,其表现介于对照组与模型组之间。TSA晚期干预组小鼠肺组织各时点蓝色胶原沉积表现与模型组相仿。4组小鼠肺组织各时点Masson染色光镜下所见见图1-4,Masson染色的IOD值见表2。
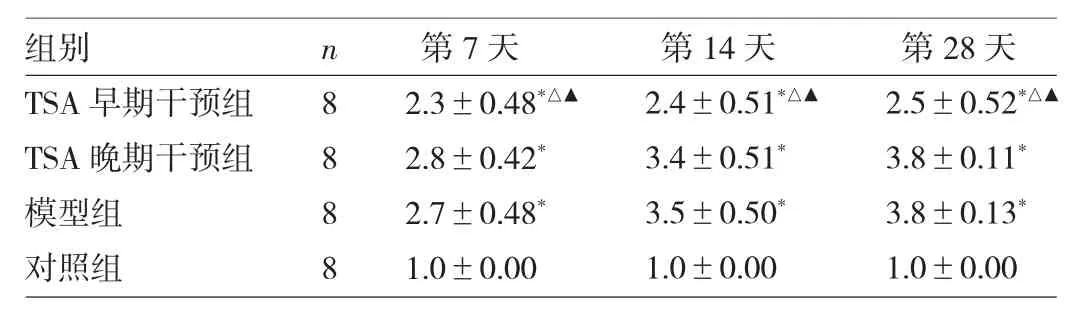
表1 4组小鼠各时点肺泡炎S z a p i e l评分比较(分)
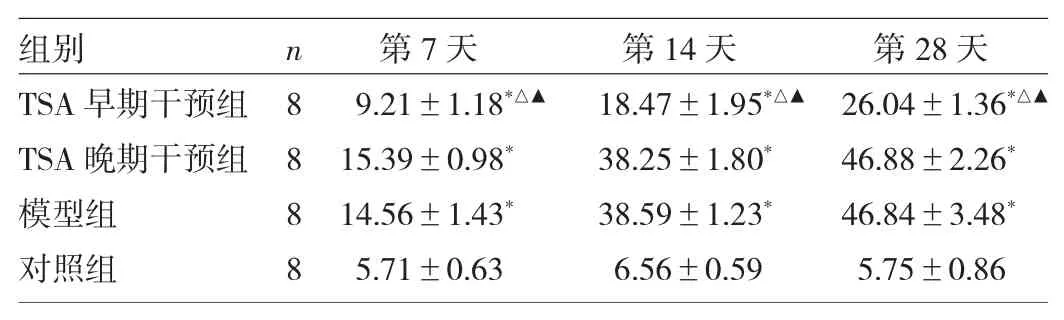
表2 4组小鼠各时点肺组织Masson染色的I OD值比较
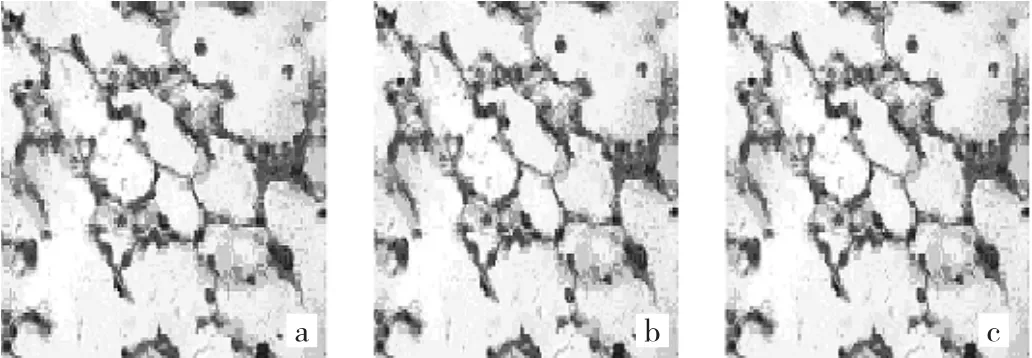
图1 对照组小鼠肺组织各时点光镜下所见(a:第7天;b:第14天;c:第28天;Masson染色,×400)
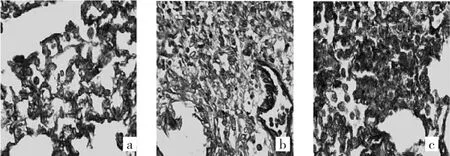
图2 模型组小鼠肺组织各时点光镜下所见(a:第7天;b:第14天;c:第28天;Masson染色,×400)
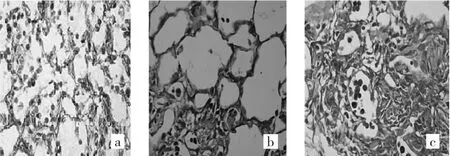
图3 T SA早期干预组小鼠肺组织各时点光镜下所见(a:第7天;b:第14天;c:第28天;Masson染色,×400)

图4 T SA晚期干预组小鼠肺组织各时点光镜下所见(a:第7天;b:第14天;c:第28天;Masson染色,×400)
由表2可见,4组小鼠肺组织各时点Masson染色的IOD值比较均有统计学差异(均P<0.05)。与对照组比较,TSA早期干预组、TSA晚期干预组、模型组小鼠肺组织各时点Masson染色IOD值均升高(均P<0.05)。与模型组比较,TSA早期干预组小鼠肺组织各时点Masson染色IOD值均降低(均P<0.05),TSA晚期干预组小鼠肺组织各时点Masson染色IOD值差异均无统计学意义(均P>0.05)。与TSA晚期干预组比较,TSA早期干预组小鼠肺组织各时点Masson染色IOD值亦均降低(均P<0.05)。
2.3 4组小鼠各时点肺组织HYP含量比较 见表3。

表3 4组小鼠各时点肺组织H Y P含量比较(m g/g肺组织)
由表3可见,4组小鼠肺组织各时点HYP含量比较均有统计学差异(均P<0.05)。与对照组比较,TSA早期干预组、TSA晚期干预组、模型组小鼠肺组织各时点HYP含量均升高(均P<0.05)。与模型组比较,TSA早期干预组小鼠肺组织各时点HYP含量均降低(均P<0.05),TSA晚期干预组小鼠肺组织各时点HYP含量差异均无统计学意义(均P>0.05)。与TSA晚期干预组比较,TSA早期干预组小鼠肺组织各时点HYP含量亦均降低(均P<0.05)。
2.4 4组小鼠各时点肺组织NF-κB表达水平比较 见表4。

表4 4组小鼠各时点肺组织N F-κB表达水平(I OD值)比较
由表4可见,4组小鼠肺组织各时点NF-κB表达水平比较均有统计学差异(均P<0.05)。与对照组比较,TSA早期干预组、TSA晚期干预组、模型组小鼠肺组织各时点NF-κB表达水平均升高(均P<0.05)。与模型组比较,TSA早期干预组小鼠肺组织各时点NF-κB表达水平均降低(均P<0.05),TSA晚期干预组小鼠肺组织各时点NF-κB表达水平差异均无统计学意义(均P>0.05)。与TSA晚期干预组比较,TSA早期干预组小鼠肺组织各时点NF-κB表达水平亦均降低(均P<0.05)。
3 讨论
肺纤维化是多种肺部慢性疾病后期的共同结局,其病理特点是早期肺泡炎和后期成纤维细胞大量增生及胶原纤维进行性沉积并取代正常的肺组织结构[9]。肺纤维化的发生与TGF-β、NF-κB等许多细胞因子有关。TGF-β通过激活下游效应分子Smads家族而介导致纤维化信号通路的传导[10]。而NF-κB通过活化转谷酰氨酶的启动子,调控多种细胞因子基因表达增加,激活TGF-β,促进成纤维细胞Ⅰ、Ⅲ型胶原的表达,介导肺泡炎和肺纤维化。NF-κB是影响肺纤维化形成与发展的重要细胞因子。
TSA为作为链霉菌代谢产物,可影响NF-κB的核转录,抑制NF-κB依赖的启动子活性[11]。研究表明,血管内皮生长因子受体抑制剂通过抑制脂多糖诱发的NF-κB激活,减少TNF-α、TGF-β等细胞因子的合成,减轻炎症因子的释放,能缓解急性肺损伤中内皮细胞炎性损害[12]。TSA通过分子靶向作用减少组蛋白的乙酰化程度,改变组蛋白乙酰化的失衡状态所引起的基因调控失衡[13],起到调控基因表达,抑制多种肿瘤细胞的生长,诱导细胞凋亡的作用[14]。研究显示,上皮细胞的过度凋亡导致肺组织结构破坏,产生肺纤维化[15]。肺纤维化时胶原纤维明显增多,HYP为胶原纤维所特有,检测肺组织HYP的含量对于判断肺纤维严重程度意义重大[16]。
本课题组前期研究发现,在小鼠肺纤维化发生过程中,肺组织HYP含量进行性增加,早期给予TSA处理后HYP含量会降低,晚期给予TSA处理则效果不明显[17]。本研究结果也同样显示早期给予TSA处理后HYP含量会降低,同时还发现TSA早期干预组小鼠肺组织NF-κB表达水平较模型组、TSA晚期干预组减少,较对照组升高。这说明肺组织NF-κB的表达水平越高,肺纤维化程度相对越严重。早期TSA干预后小鼠肺组织Masson染色显示蓝色胶原沉积明显减少,这间接说明TSA能抑制小鼠肺组织NF-κB的表达,通过早期抗炎达到抑制肺纤维化作用。
综上所述,本研究通过分析TSA对BLM诱导肺纤维化小鼠的NF-κB表达的影响,探讨防治肺纤维化的新途径。结果发现早期应用TSA或能通过抑制NF-κB的表达达到延缓肺纤维化的目的,这为目前临床肺纤维化的治疗提供重要思路。
[1]Behr J.The d iagnosis and treatment of id iopathic pulmonary fib rosis[J].Dtsch Arzteb l Int,2013,110(51-52):875-881.
[2]Ahluwalia N,Shea B S,Tager AM,etal.New therapeutic targets in id iopathic pulmonary fib rosis.Aim ing to rein in runaway woundhealing responses[J].Am JResp ir CritCare Med,2014,190(8):867-878.
[3]Kalayarasan S,Sriram N,Soumyakrishnan S,et al.Diallylsulfide attenuates excessive collagen p roduction and apop tosis in a rat model of b leomycin induced pulmonary Fib rosis through the involvement of p rotease activated recep tor-2[J].Toxicol App l Pharmacol,2013,271(2):184-195.
[4]Usuki J,Matsuda K,Azum a A,et al.Sequential analysis of m yofib rob last d ifferentiation and transform ing g row th factor-β1/ Smad pathway activation in murine pu lm onary fib rosis[J].Nippon Med Sch,2012,79(1):46-59.
[5]水华,陈德基.苯那普利对单侧输尿管梗阻大鼠肾脏的保护作用[J].中国医师杂志,2002,4(7):684-686,689.
[6]Deroanne C F,Bonjean K,Servotte S,etal.Histone deacetylases inhibitors as anti-ang iogenic agents altering vascular endothelial g row th factor signaling[J].Oncogene,2002,21(3):427-436.
[7]Maheshwari U,Gup ta D,Aggarwal A N,et al.Spectrum and d iagnosis of id iopathic pulmonary fib rosis[J].Ind ian J Chest Dis Allied Sci,2004,46(1):23-26.
[8]Szap ielSV,Elson N A,Fulmer JD,etal.Bleom yeinduce interstitial pulmonaty d isease in the nude,athym icm ouse[J].Am Rev Resp ir Dis,1979,120(4):893-899.
[9]孔勤,陈民利.特发性肺纤维化发病机制的研究进展[J].中国比较医学杂志,2012,22(8):74-74.
[10]Ham id T,Guo SZ,Kingery JR,eta l.Card iom yocyte NF-KB p65 p romotes adverse remodeling,apop tosis,and endop lasm ic reticulum stress in heart failure[J].Card iovasc Res,2011,89(1):129-138.
[11]Rahman M M,Kukita A,Kukita T,etal.Two histone deacetylase inhibitors,trichostatin A and sodium butyrate,supp ress d ifferentiation into osteoc lasts but not into mac rophages[J]. Blood,2003,101(9):3451-3459.
[12]Parekh D,DancerRC,ThickettDR.Acute lung injury[J].Clin Med (Lond),2011,11(6):615-618.
[13]Kouzarides T.Histone acetylases and deacetylases in cell p roliferation[J].CurrOp in GenetDev,1999,9(1):40-48.
[14]Yang H,Nie Y,LiY,etal.Histonemod ification-med iated CYP2E1 gene exp ression and apop tosis ofHepG2 cells[J].Exp BiolMed (Maywood),2010,235(1):32-39.
[15]DanialN N.BCL-2 fam ily p roteins:checkpoints o fapop totic cell death[J].Clin Res,2007,13(24):7254-7263.
[16]Aytem ur Z A,Hacievliyag ilSS,Iraz M,etal.Effects of ilop roston b leomycin-induced pulmonary fib rosis in rats com pared withm ethyl-p rednisolone[J].Rev PortPneumo l,2012,18(6):272-277.
[17]黄旭晴,施长春,李旭,等.曲古抑菌素A对博莱霉素致肺纤维化小鼠新生血管的影响[J].浙江医学,2014,36(5):370-373.
Trichostatin A affects expression of NF-κB in mice w ith bleomycin-induced pulmonary fibrosis
HUANG Xuqing,XU Changqing,LI Xu.Department of Respiratory Medicine,the Affiliated Hospital of Hangzhou Normal University,Hangzhou 310015,China
【 Abstract】 Objective To investigate the effect of trichostatin A on exp ression of NF-κB in m ice w ith b leomycin-induced pulmonary fibrosis. Methods Ninty-six male C57BL/6 m ice were random ly allocated into four g roups w ith 24 in each g roup:saline controlg roup(control g roup),b leomycin-induced pulmonary fib rosis g roup(model g roup),early TSA treatment group(early g roup),and late TSA treatmentg roup(late g roup).Eightm ice in each g roup were sacrificed at d7,d14 and d28.The lung tissue samp les were taken and hematoxylin eosin and Masson staining were adop ted to evaluate the severity of pulmonary inflammation and fib rosis.The contents of hyd roxyp ro1ine(HYP)in the lung tissues were detected by alkaline hyd rolysis technique. Results The lung tissue in model group p resented significant b leed ing and effusion at d7 and pulmonary fib rosis at d28.Bleed ing,effusion and pulmonary fib rosis were attenuated in early treatment g roup com pared to model group at the same time points.Hyd roxyp roline content and the exp ression of NF-κB in lung tissue were significantly higher in model g roup than those in control g roup(both P<0.05).Compared w ith model g roup,early g roup showed m ilder alveolitis and lower Szapielscore of pulmonary fib rosis,decreased hyd roxyp rollne content and NF-κB exp ression at the same time points(both P<0.05).However,there were no significant differences in above ind icators between late g roup and model group. Conclusion Early treatmentof TSA can alleviate b leom ycin-induced pulmonary fibrosis via inhibiting early inflammation and inhibiting exp ressions of NF-κB.
Trichostatin A Pulmonary fib rosis NF-κB
2016-05-23)
(本文编辑:李媚)
杭州市社会发展自主申报项目(20160533B18);浙江省公益性技术应用研究计划项目(2015C33258);杭州市卫生科技计划项目(2011A017)
310015 杭州师范大学附属医院呼吸科
黄旭晴,E-mail:h x q3871@126.com
猜你喜欢
中老年保健(2022年2期)2022-11-25
昆明医科大学学报(2022年4期)2022-05-23
昆明医科大学学报(2021年4期)2021-07-23
东方考古(2021年0期)2021-07-22
摄影之友(影像视觉)(2020年2期)2021-01-14
渔业研究(2018年5期)2018-01-17
医学美学美容·中旬刊(2015年2期)2015-10-21
中国医疗美容(2015年4期)2015-04-27
郑州大学学报(医学版)(2015年2期)2015-02-27
体育科研(2013年2期)2013-05-31
