
(Pr0.9La0.1)2(Ni0.74Cu0.21Ga0.05)O4+δ-Ce0.9Gd0.1O2-δ复合阴极的制备及电化学性质
2016-12-20 02:21盛双赵辉郝举红孙丽萍霍丽华
无机化学学报 2016年12期
盛双 赵辉*, 郝举红 孙丽萍*, 霍丽华
(Pr0.9La0.1)2(Ni0.74Cu0.21Ga0.05)O4+δ-Ce0.9Gd0.1O2-δ复合阴极的制备及电化学性质
盛双1赵辉*,1郝举红2孙丽萍*,1霍丽华1
(1黑龙江大学化学化工与材料学院,功能无机材料化学教育部重点实验室,哈尔滨150080)
(2哈尔滨远东理工学院,哈尔滨150025)
采用EDTA-柠檬酸盐法制备了(Pr0.9La0.1)2(Ni0.74Cu0.21Ga0.05)O4+δ(PLNCG),并与Ce0.9Gd0.1O2-δ(CGO)形成复合阴极PLNCG-CGO。XRD和SEM分析结果表明PLNCG与CGO在1 000℃具有较好的化学相容性。电化学阻抗测试结果表明PLNCG-30%CGO复合阴极在700℃的极化电阻为0.092Ω·cm2;过电位为39.3 mV时,电流密度达到113.3 mA·cm-2。氧分压分析表明电极反应的速率控制步骤为电荷转移过程。阳极支撑单电池(Ni-CGO/CGO/PLNCG-30%CGO)在700℃的最大输出功率密度达到569 mW· cm-2,开路电压(OCV)为0.76 V。
中温固体氧化物燃料电池(IT-SOFCs);复合阴极;PLNCG-CGO;电极反应
固体氧化物燃料电池(SOFC)能够将化学能直接转化为电能,具有能量转化效率高,环境友好等诸多优点,是最具发展前景的能源转换装置之一[1]。SOFC较高的工作温度(通常大于900℃)常常导致一系列问题,实现工作温度的中低温化具有重要意义。作为重要组件之一,SOFC阴极性能的优劣直接影响电池的输出性能。伴随操作温度的降低,传统阴极材料的极化电阻急剧增大,因此开发中低温(500~700℃)下具有较高催化活性的新型阴极材料成为研究热点[2-7]。
钴基氧化物La0.6Sr0.4Co0.8Fe0.2O3-δ[8]、Ba1-xSrxFe1-yCoyO3-δ[9-10]、SrCo1-xNbxO3-δ[11]等在中低温下具有较低的极化电阻及较高的电催化活性,作为中温固体氧化物燃料电池(IT-SOFC)阴极材料已得到广泛研究。然而钴基材料较大的热膨胀系数及较高制备成本等缺点制约其大规模应用[10]。近来研究表明Pr2NiO4具有较好的电催化活性[13-14],但是其结构稳定性较差。研究发现Pr位掺杂La,Ni位掺杂Cu能有效改善其高温相稳定性[15-17]。Ishihara等发现在Pr2(Ni0.75Cu0.25)O4+δ中引入d10结构的Ga3+能产生更多的间隙氧,进而增强其氧渗透性[18]。Peng等进一步合成了(Pr0.9La0.1)2(Ni0.74Cu0.21Ga0.05)O4+δ(PLNCG),证实其作为SOFC阴极材料具有潜在的研究价值[19]。
大量研究结果表明,在阴极材料中引入离子导电相(如CGO)可以有效降低阴极过电位,提升电化学性能[20-24]。这是因为对于电子电导为主的阴极材料,其离子电导率较低,氧还原反应一般仅限于电极、电解质及空气的三相接触界面;而对于离子-电子混合导电的阴极材料,虽然三相界面反应区会向阴极内部扩展,但也仅限于电极/电解质界面附近较窄的范围。因此在电极材料中加入离子导电相,电极反应区域将扩展到整个阴极内部,同时CGO的高氧离子电导率能够有效改善复合电极的氧离子传导能力。迄今为止,PLNCG与离子导电材料形成复合电极的研究还未见报道。考虑到PLNCG较好的阴极性能,本文将其与Ce0.9Gd0.1O2-δ(CGO)材料进行复合,进一步改善电极的氧催化还原性能,并探索PLNCG电极的电化学反应机理。
1 实验部分
1.1PLNCG和CGO粉体的制备
采用EDTA-柠檬酸盐法制备PLNCG材料。准确称取分析纯的Pr6O11、La2O3、NiO、Ga2O3、CuO溶于2 mol·L-1硝酸中,配置成金属离子浓度为0.5 mol· L-1的硝酸盐溶液。按照化学计量配比nPr:nLa:nNi:nCu: nGa=1.8:0.2:0.74:0.21:0.05准确移取上述溶液,再按照总的金属离子、EDTA、柠檬酸的物质的量比例为1:1:2加入EDTA与柠檬酸,搅拌使其完全溶解。加入氨水调节溶液的pH值为7,80℃搅拌直至生成暗绿色凝胶,180℃烘箱中干燥,950℃煅烧10 h得到PLNCG粉体。CGO粉体的制备参照文献[25]。
1.2测试电池的制备
将CGO粉体在220 MPa加压成型,1 200℃烧结12 h,1 400℃烧结24 h,得到致密度约为95%的电解质圆片。将PLNCG与CGO粉末按照不同质量百分比例混合(表示为PLNCG-x%CGO,x= 0,10,20,30,40),加入适量松油醇(含3%乙基纤维素),充分研磨使其混合均匀。将得到的浆料对称涂在CGO电解质圆片的两侧,然后在空气中不同温度(900~1 100℃)下烧结2 h制成对称电极半电池。
三电极半电池的制备:将上述浆料均匀地涂在CGO电解质基片的一侧,作为工作电极(WE);另一侧对称地涂上Pt浆作为对电极(CE)。Pt丝粘结在工作电极一侧作为参比电极(RE),1 000℃烧结2 h。
称取一定质量阳极粉(质量比mNiO:mCGO=6:4)和CGO电解质粉末,采取双粉干压法在120 MPa下压制成直径为15 mm的阳极支撑半电池素坯,1 350℃烧结4 h。再将阴极浆料均匀涂在电解质薄膜上,1000℃烧结2 h,得到阳极支撑型单电池(Ni-CGO/ CGO/PLNCG-CGO)。
1.3材料的测试及表征
粉体材料的物相采用Bruker D8-Advance型X射线粉末衍射仪进行表征分析。射线管电压为40 kV,工作电流10 mA,射线源为Cu靶Kα射线(λ= 0.154 18 nm)。电极的微观形貌采用HITACHI S-4800型扫描电子显微电镜(SEM)进行观测。电极的电化学性能用交流阻抗技术进行研究(Autolab PGStat30电化学工作站),频率范围10-2Hz~1 MHz,温度范围500~700℃,空气与N2的混合气氛。阴极极化曲线采用电压阶梯扫描技术测试,并利用公式ηWE=ΔUWR-iRel计算阴极极化过电位,其中ηWE为阴极过电位,ΔUWR为工作电极与参比电极的电位差,i为流经电池的电流,Rel为电解质电阻。采用Biologic VSP(SN 0315)装置测试阳极支撑单电池的输出性能,以加湿的H2(含体积分数约3%的H2O)为燃料气,空气为氧化气,测试温度600~700℃。
2 结果与讨论
2.1材料的物相及高温化学稳定性
图1(a)为PLNCG粉体室温下的XRD图、Rietveld精修结果、二者的差值及Bragg衍射峰位置。可见所有的衍射峰均归属为PLNCG的衍射峰,没有其他杂相生成。精修结果显示PLNCG为四方晶系,空间群为I4/mmm;计算得到的晶胞参数:a=b=0.383 23 nm,c=1.255 26 nm,Rp=8.72%,Rwp= 10.83%。Rietveld精修结果与文献[26]报道的结果一致,表明所合成的材料为单一的PLNCG物相。
为了考察PLNCG与CGO电解质材料的高温化学稳定性,将两种粉体按照质量百分比为7:3的比例进行混合,在1 000℃空气中煅烧10 h。图1(b)为混合粉体煅烧后的XRD图、Rietveld精修结果、二者的差值及Bragg衍射峰位置。计算得到PLNCG的晶胞参数为:a=b=0.382 99 nm,c=1.253 92 nm,电解质CGO的晶胞参数为a=b=c=0.541 19 nm,Rp=9.69 %;Rwp=11.73%,与标准数据对比并未发现有明显改变,且在图中未发现其他杂峰。表明PLNCG-CGO混合粉体在1 000℃未发生化学反应,两者之间具有较好的高温化学相容性。
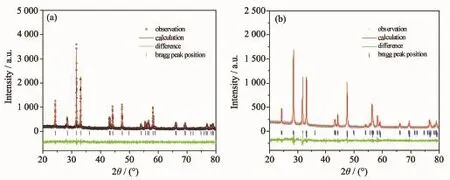
图1 (a)PLNCG,(b)PLNCG-CGO在1000℃煅烧10 h后粉体的XRD图,Rietveld精修结果、二者的差值及Bragg衍射峰位置Fig.1 Experimental and calculated XRD patterns and the difference between them,calculated Bragg peaks positions for(a) PLNCG and(b)PLNCG-CGO mixed powders after sintered at 1 000℃for 10 h
2.2电极微观结构表征
电极的烧结情况直接影响其性能的优化。图2为PLNCG-30%CGO复合阴极表面及横断面的SEM图。可见当电极的烧结温度为900℃时,电极粒子之间的烧结连接不够充分(图2(a));而1 100℃烧结后的电极表面,粒子之间明显发生了熔融、团聚现象(图2(c))。图2(b)为PLNCG-30%CGO复合阴极在1 000℃烧结后的表面SEM图。可以观察到PLNCG与CGO粒子之间存在一定的烧结连接,且电极内部形成了较好的孔隙结构,这有利于电荷的传输和气体扩散。电极材料与电解质之间形成良好接触,电极厚度约为20μm(图2(d))。

图2 不同烧结温度下PLNCG-30%CGO复合阴极表面(a)900℃(b)1 000℃(c)1 100℃及横断面(d)1 000℃的SEM图Fig.2 SEM images of PLNCG-30%CGO composite electrode sintered at various temperatures,surface(a)900℃(b)1 000℃(c)1 100℃and cross-section(d)1 000℃
2.3电化学表征
为了进一步验证烧结温度对电极性能的影响,测试了不同温度烧结的复合电极在700℃空气中的阻抗谱(图3)。可见阻抗谱均由2个弧构成。采用图3所示等效电路对EIS数据进行拟合分析,其中Rel为电解质及导线的欧姆电阻,QH与QL分别为2个常相位元件,RH、RL分别代表高频弧与低频弧对应的极化电阻值,总的极化电阻RP=RH+RL。由拟合结果可见电极烧结温度对于RH与RL均有一定的影响(图3)。当烧结温度由900℃升高到1 000℃时,RH值降低了30%,RL值变化并不明显;而当烧结温度继续上升到1 100℃时,RL值显著增大,且在低频区出现明显半圆弧。由此确定电极的最佳烧结温度为1 000℃。电极烧结温度对于极化电阻产生影响的主要原因,可能是由于900℃烧结的电极粒子之间连接不良,导致电荷在传导过程中受到阻碍;而温度过高导致粒子团聚,电极有效面积减少。阻抗的分析结果与SEM测试结果相符(图2)。类似的烧结温度影响电极极化电阻的现象在其他文献也有报道[27-29]。

图3 不同温度条件下烧结2 h的PLNCG-30%CGO复合电极在700℃空气中测得的阻抗谱Fig.3 Impedance spectra of PLNCG-30%CGO electrode sintered at different temperatures for 2 h and then measured at 700℃in air
CGO的相对含量对复合阴极的极化电阻有较大的影响。图4为空气中700℃测得的不同含量CGO复合阴极的阻抗谱图。采用图3所示等效电路进行拟合分析,发现复合阴极的极化电阻随着CGO复合量的增加而逐渐减小,且主要表现在高频区域。当CGO复合量从0增加到30%,RH从0.189Ω·cm2降低到0.088Ω·cm2,降低了约53.44%,RP值也由0.197Ω·cm2降低到0.092Ω·cm2。这种性能的提高可以解释为:具有离子导电性的CGO改善了复合电极中的氧传导路径,扩大了电化学反应区域,使氧还原反应由电极与电解质交界处扩散到了整个阴极层[30]。而当复合量为40%时,电极的极化电阻反而变大(RP=0.136Ω·cm2)。这主要是由于CGO的加入量过多,电子的传输路径受到阻塞,进而使电极的导电性降低,极化电阻增大[31]。
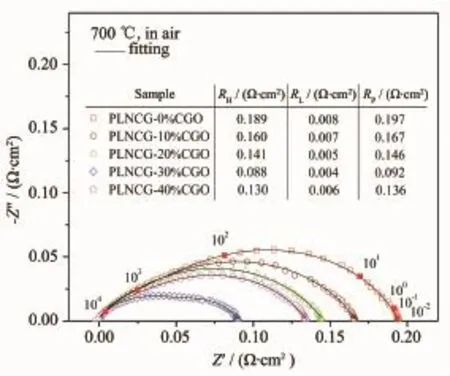
图4 PLNCG-x%CGO(x=0,10,20,30,40)阴极在空气中700℃测得的阻抗谱图Fig.4 Impedance spectra of PLNCG-x%CGO(x= 0,10,20,30,40)cathode measured at 700℃in air
为了研究电极上的反应动力学过程,本文测试了700℃不同氧分压下PLNCG-30%CGO复合阴极的阻抗谱图。由图5a可见,不同氧分压测试的阻抗谱均由2个曲线弧组成,且高频弧对应的阻抗值大于低频弧,说明高频区所对应的电化学反应过程为电极反应的速控步骤。采用图3中所示的等效电路对得到的阻抗谱进行拟合分析,并根据以下公式计算每一个曲线弧所对应的电容值和弛豫频率f:

式中Q为常相位元件,n代表与真实电容的相似程度(n=1时,Q=C),R为曲线弧对应的阻抗数值[32]。由Q、n和R计算出高频弧(频率104~102Hz)对应的电容值为10-4F·cm-2(表1)。根据文献可知,这种典型的电容值涉及的是氧原子在电极表面上的电荷转移反应(charge transfer reaction)[33-34];低频弧(频率低于102Hz)对应的电容值为10-1F·cm-2,为氧分子的吸附、扩散过程[32]。作为对比,在相同实验条件下测试了PLNCG阴极的电化学交流阻抗(图5(b))。发现PLNCG阴极在不同氧分压下测试的阻抗谱图也是由2个曲线弧构成,拟合结果显示高频弧(频率104~102Hz)对应电容值为10-4F·cm-2(表2),低频弧(频率低于102Hz)对应的电容值为10-1F·cm-2,与复合阴极的电容值一致,表明CGO的加入并未改变PLNCG阴极的电化学反应机理。另外对比表1和表2的拟合数据可见,同一氧分压下PLNCG-30%CGO复合阴极的RP值均小于PLNCG阴极,这一结果进一步说明复合CGO后,电极上的氧还原反应活性位点增加,氧的传导路径得到改善,促进了电极上的电化学反应过程。
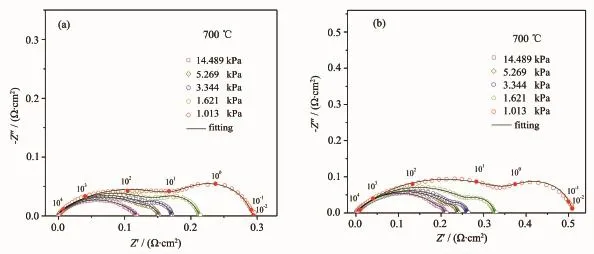
图5 700℃不同氧分压(a)PLNCG-30%CGO(b)PLNCG的阻抗谱图Fig.5 Impedance spectra for(a)PLNCG-30%CGO and(b)PLNCG at 700℃under various oxygen partial pressures
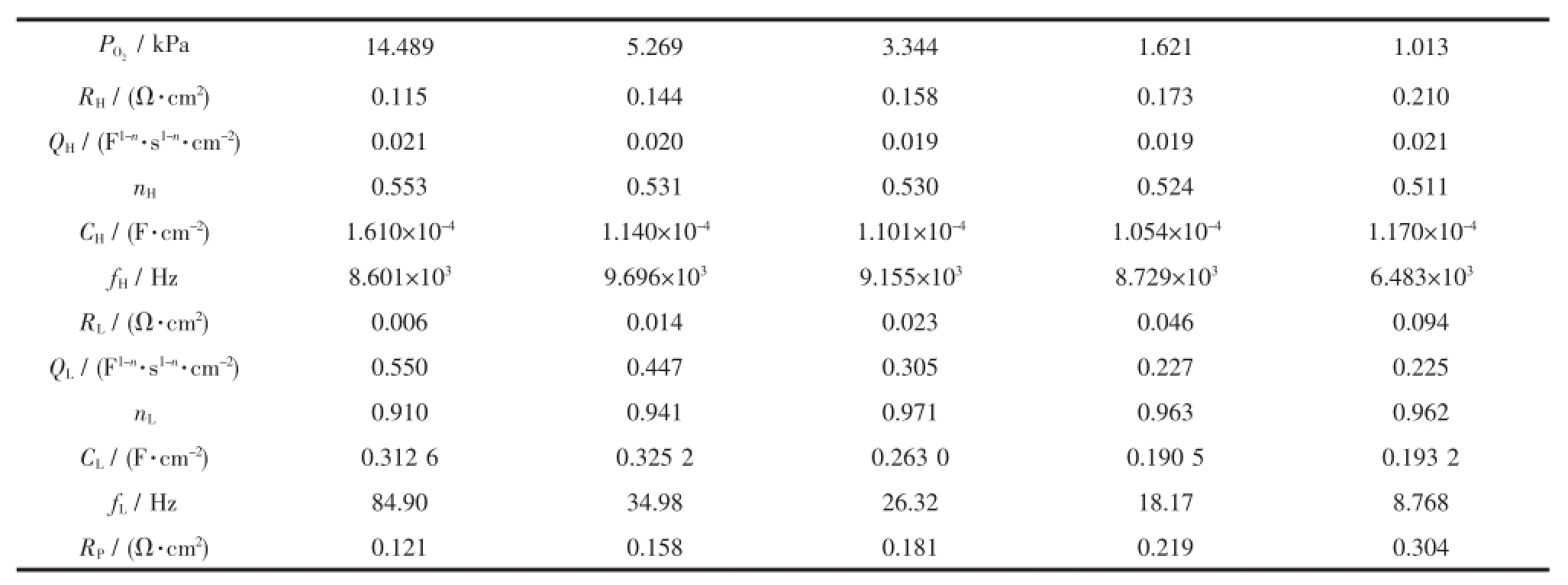
表1 700℃不同氧分压下PLNCG-30%CGO复合阴极阻抗谱拟合后的RH、RL及对应的电容值与弛豫频率Table 1 Fitted impedance spectra values RH,RL,capacitance,and relaxation frequency of composite cathode PLNCG-30% CGO at 700℃under various oxygen partial pressures

表2 700℃不同氧分压下PLNCG阴极阻抗谱拟合后的RH、RL及对应的电容值与弛豫频率Table 2 Fitted impedance spectra values RH,RL,capacitance,and relaxation frequency of cathode PLNCG at 700℃under various oxygen partial pressures
为了进一步讨论复合电极上的电化学反应机理,我们将极化电阻与对应氧分压作图(图6)。由图可见极化电阻与氧分压的变化关系可以通过以下公式来进行描述:

式中的n值反映了电极上发生的电化学反应类型[35-36]。
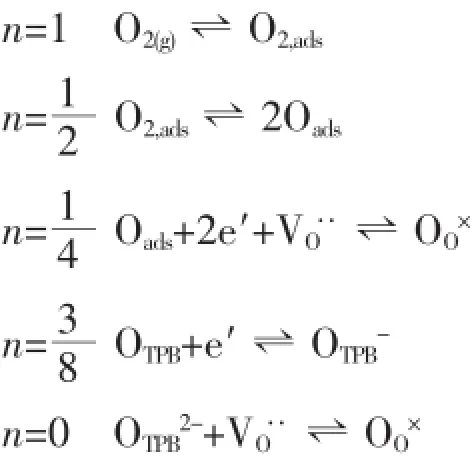

图6 700℃阴极极化电阻随氧分压的变化曲线Fig.6 Curves of cathode polarization resistance with oxygen partial pressures at 700℃
图7为空气中不同温度下测得的复合阴极的电流-电压极化曲线。由图可知,当电流密度相同时,该复合阴极的过电位随着温度的升高而逐渐降低。在700℃条件下,过电位为39.3 mV时,PLNCG-30%CGO复合阴极的电流密度可达到113.3 mA· cm-2,优于文献报道的一些非钴系阴极材料,如SrFe0.7Cu0.3O3-δ(ηWE=90 mV时,i=103 mA·cm-2)[37]、Pr2NiMnO6(ηWE=37 mV时,i=102 mA·cm-2)[40]。
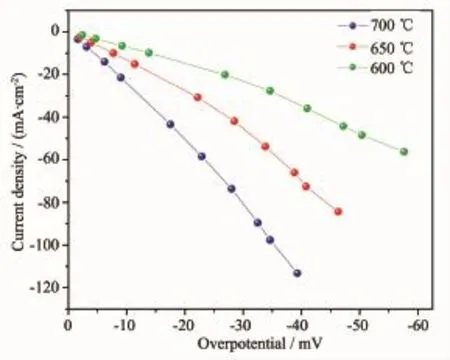
图7在空气中不同温度下PLNCG-30%CGO复合阴极的过电位-电流密度曲线Fig.7 Overpotential-current density curves for PLNCG-30%CGO composite cathode measured at different temperatures in air
图8为阳极支撑NiO-CGO/CGO/PLNCG-30% CGO单电池的I-V-P曲线。单电池的开路电压(OCV)随温度的升高而减小,与文献报道的变化趋势一致[41-42]。由于高温还原气氛下电解质CGO中的Ce4+部分还原成Ce3+,电子载流子浓度增加,导致电子导电性增强,OCV下降。700℃时,开路电压为0.76 V,最大功率密度达到569 mW·cm-2,优于阳极支撑的单电池NiO-YSZ/YSZ/PLNCG(506 mW·cm-2)[19],以及金属支撑的以La0.8Sr0.2MnO3-δ/YSZ为复合阴极构筑的单电池(800℃最大功率密度为0.38 W·cm-2)[43]。以上结果表明PLNCG-30%CGO复合阴极有较高的电极催化活性,是一种很有发展前景的固体氧化物燃料电池阴极材料。
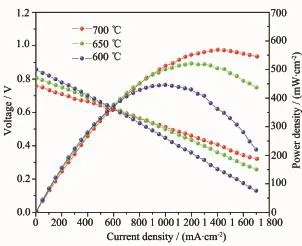
图8 不同温度下NiO-CGO/CGO/PLNCG-30%CGO单电池的I-V-P曲线Fig.8 I-V-P curves of single cell NiO-CGO/CGO/PLNCG-30%CGO at different temperatures
3 结论
采用EDTA-柠檬酸盐法合成了PLNCG粉体材料,并制备了PLNCG-CGO复合阴极。研究发现,CGO的引入增加了电极上的氧还原反应活性位点,改善了氧的传导路径,促进了电极上的电荷转移反应过程。PLNCG-30%CGO复合阴极的极化电阻700℃为0.092Ω·cm2,阳极支撑单电池的输出功率密度达到569 mW·cm-2。PLNCG-30%CGO复合阴极是一种潜在的IT-SOFC阴极材料。
[1]Steele B C H,Heinzel A.Nature,2001,414:345-352
[2]BeckelD,Bieberle-Hütter A,Harvey A,etal.J.Power Sources, 2007,173(1):325-345
[3]SUN Li-Ping(孙丽萍),LI Qiang(李强),ZHAO Hui(赵辉), et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(5): 1045-1050
[4]Li Q,Sun L P,Huo L H,et al.Int.J.Hydrogen Energy, 2010,35:9151-9157
[5]Xia T,Lin N,Zhao H,et al.J.Power Sources,2009,192: 291-296
[6]ZHANG Han(张瀚),XIA Chang-Rong(夏长荣).Chinese J. Inorg.Chem.(无机化学学报),2010,26(10):1875-1879
[7]XIAO Hui(肖辉),SUN Li-Ping(孙丽萍),ZHAO Hui(赵辉), et al.Chinese J.Inorg.Chem.(无机化学学报),2015,31(6): 1139-1144
[8]Xu Q,Huang D P,Zhang F,et al.J.Alloys Compd.,2008, 454:460-465
[9]Liu Z,Cheng L Z,Han M F.J.Power Sources,2011,196: 868-871
[10]Shao Z P,Haile S M.Nature,2004,431:170-173
[11]Wang F,Zhou Q J,He T M,et al.J.Power Sources,2010, 195:3772-3778
[12]Ding H P,Lin B,Liu X Q,et al.Electrochem.Commun., 2008,10:1388-1391
[13]Zhou X D,Templeton J W,Nie Z,et al.Electrochim.Acta, 2012,71:44-49
[14]Ferchaud C,Grenier J C,Ye Z S,et al.J.Power Sources, 2011,196:1872-1879
[15]Yashima M,Yamada H,Ishihara T,et al.Chem.Mater., 2012,24:4100-4113
[16]Wang Y F,Cheng J G,Jiang Q M,et al.J.Power Sources, 2011,196:3104-3108
[17]Kovalevsky A V,Kharton V V,Yaremchenko A A,et al.J. Electroceram,2007,18:205-218
[18]Ishihara T,Nakashima K,Okada S,et al.Solid State Ionics, 2008,179:1367-1371
[19]Peng S J,Wei Y Y,Xue J,et al.Int.J.Hydrogen Energy, 2013,38:10552-10558
[20]Zhou Q J,Wang F,Shen Y,et al.J.Power Sources,2010, 195:2174-2181
[21]LI Qiang(李强),FAN Yong(范勇),SUN Li-Ping(孙丽萍), et al.Chinese J.Inorg.Chem.(无机化学学报),2007,23(2): 300-304
[22]Leng Y J,Chan S H,Liu Q L.Int.J.Hydrogen Energy, 2008,33:3808-3817
[23]Park Y M,Kim J H,Kim H.Int.J.Hydrogen Energy,2011, 36:9169-9179
[24]Khandale A P,Lajurkar R P,Bhoga S S.Int.J.Hydrogen Energy,2014,39:19039-19050
[25]Meng X W,LüS Q,Ji Y,et al.Ceram.Int.,2015,41:12107 -12114
[26]Yashima M,Sirikanda N,Ishihara T.J.Am.Chem.Soc., 2010,132:2385-2392
[27]Li Q,Fan Y,Zhao H,et al.J.Power Sources,2007,167:64-68
[28]Sun C,Li Q,Sun L P,et al.Mater.Res.Bull.,2014,53:65-69
[29]Li Q,Zhao H,Huo L H,et al.Electrochem.Commun., 2007,9:1508-1512
[30]Dusastre V,Kilner J A.Solid State Ionics,1999,126:163-174
[31]Simner S P,Anderson M D,Coleman J E,et al.J.Power Sources,2006,161:115-122
[32]Martínez J P,López D M,Morales R,et al.Int.J.Hydrogen Energy,2009,34:9486-9495
[33]Pang S L,Jiang X N,Li X N,et al.J.Power Sources, 2012,204:53-59
[34]Chen D J,Ran R,Zhang K,et al.J.Power Sources,2009, 188:96-105
[35]Gao Z,Liu X M,Bergman B,et al.J.Power Sources,2011, 196:9195-9203
[36]Zhao H,Huo L H,Gao S.J.Power Sources,2004,125:149-154
[37]Li Q,Xia T,Sun L P,et al.Electrochim.Acta,2014,150: 151-156
[38]Li J,Zhang N Q,Ni D,et al.Int.J.Hydrogen Energy, 2011,36:7641-7648
[39]Lee H H,Park I Y,Park J H,et al.Int.J.Hydrogen Energy,2015,40:11998-12002
[40]Li H,Sun L P,Li Q,et al.Int.J.Hydrogen Energy,2015, 40:12761-12769
[41]Zhang L L,Liu M,Huang J H,et al.Int.J.Hydrogen Energy,2014,39:7972-7979
[42]Wang Y X,Zhao X Y,LüS Q,et al.Ceram.Int.,2014,40: 7321-7327
[43]Baek S W,Jeong J,Schlegl H,et al.Ceram.Int.,2016,42: 2402-2409
Preparation and Electrochemical Properties of(Pr0.9La0.1)2(Ni0.74Cu0.21Ga0.05)O4+δ-Ce0.9Gd0.1O2-δComposite Cathode
SHENG Shuang1ZHAO Hui*,1HAO Ju-Hong2SUN Li-Ping*,1HUO Li-Hua1
(1Key Laboratory of Functional Inorganic Material Chemistry,Ministry of Education,School of Chemistry and Materials Science,Heilongjiang University,Harbin 150080,China)
(2Harbin Far East Institute of Technology,Harbin 150025,China)
(Pr0.9La0.1)2(Ni0.74Cu0.21Ga0.05)O4+δ(PLNCG)is prepared by EDTA-citrate process,and forms composite cathode with Ce0.9Gd0.1O2-δ(CGO).XRD and SEM results demonstrate that PLNCG is chemicalcompatible with CGO at 1 000℃.The electrochemicalmeasurements show thatthe polarization resistance(Rp)of PLNCG-30%CGO is 0.092Ω·cm2at700℃.When the currentdensity reaches 113.3 mA·cm-2,the cathode overpotentialis only 39.3 mV at700℃in air.The oxygen partialpressure dependence of Rpindicates thatthe rate limiting step ofthe composite cathode is charge transferprocess.The maximum powerdensity ofanode-supportsingle cell(Ni-CGO/CGO/PLNCG-30%CGO)reaches 569 mW·cm-2at700℃with open circuitvoltage(OCV)of0.76 V.
intermediate temperature solid oxide fuel cells(IT-SOFCs);composite cathode;PLNCG-CGO;electrode reaction
O614.33;O614.121;O614.81+3
A
1001-4861(2016)12-2143-08
10.11862/CJIC.2016.275
2016-07-08。收修改稿日期:2016-10-14。
国家自然科学基金(No.51302069,51372073)、高等学校博士学科点专项科研基金(No.20132301110002)、黑龙江省自然科学基金(No. E2016051)和人事部留学人员科技活动择优资助项目(No.2014-240)资助。
*通信联系人。E-mail:lipingsun98@yahoo.com
猜你喜欢
中国粉体技术(2022年5期)2022-09-06
有色设备(2022年2期)2022-08-06
中国粉体技术(2022年2期)2022-03-19
粉末冶金技术(2021年1期)2021-03-29
粉末冶金技术(2021年1期)2021-03-29
军民两用技术与产品(2021年10期)2021-03-16
医疗装备(2020年9期)2020-05-28
电子制作(2018年12期)2018-08-01
教育教学论坛(2018年24期)2018-07-24
