
分子动力学模拟汽油组分在不同脱硫膜材料中的扩散行为
2016-12-16 03:08侯影飞刘熠斌牛青山
石油学报(石油加工) 2016年6期
侯影飞, 蒋 驰, 刘 敏, 刘熠斌, 李 鹏, 牛青山
(1.中国石油大学 重质油国家重点实验室 化学工程学院, 山东 青岛 266580;2.新疆应用职业技术学院 石油与化学工程系, 新疆 奎屯 833200)
分子动力学模拟汽油组分在不同脱硫膜材料中的扩散行为
侯影飞1,2, 蒋 驰1, 刘 敏1, 刘熠斌1, 李 鹏1, 牛青山1
(1.中国石油大学 重质油国家重点实验室 化学工程学院, 山东 青岛 266580;2.新疆应用职业技术学院 石油与化学工程系, 新疆 奎屯 833200)
为探究渗透气化脱硫膜材料脱硫性能的决定因素,采用分子动力学(MD)模拟了典型汽油组分(噻吩、正庚烷、环己烷、环己烯和甲苯)在乙基纤维素(EC)、聚乙二醇(PEG)和聚二甲基硅氧烷(PDMS)中扩散过程,绘制小分子在聚合物中扩散轨迹并分析了其扩散方式。计算求得不同分子在不同膜材料中的扩散速率。通过分析扩散分子与聚合物体系的相互作用能、扩散分子三维尺寸、聚合物自由体积和聚合物链段运动对分子扩散速率的影响,考察了分子在聚合物中的微观扩散机理。结果表明,在3种膜材料中噻吩的扩散速率均大于其余4组分,与实验结果相一致;扩散分子与聚合物相互作用是噻吩类硫化物在脱硫膜材料中优先扩散的决定因素,且其余3种因素相互协同同样影响着其在聚合物中的扩散速率。
分子动力学; 分子扩散; 汽油脱硫; 膜分离
随着世界各国对环境保护的日益重视,以及环保法规的日益严格,生产低硫及超低硫汽油已成迫切需求[1]。FCC汽油是汽油的主要来源,其所含的硫化物以硫醇、硫醚、二硫化物和噻吩类这4种有机硫化物为主,其中噻吩类硫含量占总硫含量的60%以上,因此,FCC汽油脱硫主要以脱除噻吩硫为主[2]。与常规加氢脱硫技术相比,渗透气化汽油脱硫技术具有投资和操作费用低、可深度脱硫、辛烷值损失小等优点,在实验室和工业应用层面都取得了长足的进展[3]。目前,有关渗透气化汽油脱硫技术的研究主要集中在对膜材料的选择、评价和工艺开发,对汽油组分在膜材料中的微观扩散行为和机理以及硫化物在脱硫膜材料中优先扩散的决定因素的研究较少。
随着分子模拟技术的发展,其已成为研究高分子膜材料的分子结构以及渗透组分在膜中溶解扩散过程的重要工具[4]。分子动力学(MD)可以在分子水平上精确计算小分子在高分子膜中的扩散性。Li等[5]采用分子动力学模拟研究了苯和水在聚二甲基硅氧烷(PDMS)和杯[4]芳烃(CA)共混膜中的扩散行为,分析了不同共混比例膜体系中PDMS和CA相互作用能、聚合物链段运动性、膜材料自由体积,并计算了苯和水在共混体系中的扩散系数。结果表明,分子扩散速率不仅与膜自由体积有关,而且受到CA与扩散分子的相互作用力的影响,模拟结果与实验相符。Ling等[6]采用分子动力学模拟了甲基萘和二苯并噻吩在不同膜材料中的扩散速率,结果与实验相一致;考察了影响分子扩散速率的因素,结果表明,扩散分子与膜材料相互作用能是影响分子扩散速率的最主要因素。Amani等[7]采用分子动力学模拟研究了不同氨基甲酸酯基/脲基比例的聚氨酯-聚脲(PUUs)膜的纳米结构,并考察了 O2、N2、CO2、CH4、H2S气体分子在不同PUUs膜中的分离性能,结果与实验相一致。然而到目前为止,对多汽油组分模型在聚合物中扩散行为的分子模拟研究鲜见报道。
笔者选用乙基纤维素(EC)、聚乙二醇(PEG)和聚二甲基硅氧烷(PDMS)3种常用于汽油膜分离脱硫研究的材料[8],采用分子动力学模拟技术研究噻吩、正庚烷、环己烷、环己烯和甲苯5种汽油组分在不同膜材料中的扩散行为,观察分子的扩散方式,并通过Einstein方程计算汽油组分在不同聚合物中的扩散速率;通过考察扩散分子与不同聚合物体系的相互作用、分子三维尺寸、聚合物自由体积和聚合物分子链段运动性,探究影响分子在聚合物中扩散性的因素。
1 汽油组分在膜材料中扩散行为的模拟方法与模型构建
分子动力学方法是以经典力学为基础的模拟计算方法。建立合适体系后,对体系中每个原子求解牛顿运动方程,计算各原子受力及加速度来确定每个时刻各原子的位置及其速度,进而通过体系运动轨迹计算宏观物理性质[6]。笔者使用Materials Studio的Amorphous Cell和Forcite模块进行分子动力学模拟。模拟过程中使用COMPASS力场,体系氢键计算使用DREIDING力场,能量最小化使用Smart方法,静电相互作用力计算选择Ewald方法,范德华作用力选择Atom-based方法[6]。
分别以EC、PEG和PDMS与5种典型汽油组分混合构建聚合物体系。不同汽油组分按照实验常用模拟汽油比例[9]填充,且在3种聚合物体系中汽油组分占总质量的15%。按照初始密度0.6 g/cm3构建10个晶胞结构并进行优化,选取能量最低结构,在298 K、101.325 kPa下进行200 ps的NPT系综动力学模拟,使体系接近真实密度;然后对体系进行200 ps的NVT系综动力学模拟使结构弛豫,再对其在101.325 kPa下进行500 ps的NPT系综退火循环加速体系平衡,温度从298 K到498 K,温度梯度为25 K。退火后的结构几何优化后进行500 ps的NPT系综动力学模拟,使体系达到298 K下真实密度,晶胞结构如图1所示,分子模型采用球棍结构;继续升温到330.15 K,依次进行500 ps的NPT动力学模拟和500 ps的NVT系综动力学模拟,使体系在目标温度下充分平衡,然后进行2500 ps的NVT动力学计算,模拟汽油组分扩散过程。模拟数据列于表1。

图1 3种聚合物体系的分子模型

表1 3种聚合物体系分子模拟数据
2 结果与讨论
2.1 小分子在聚合物中的扩散方式
笔者以噻吩作为扩散分子,通过Materials Studio脚本分析动力学轨迹文件,获得噻吩分子每一帧的重心坐标,绘制出噻吩分子在一个聚合物晶胞体积内的运动轨迹,如图2所示。模拟结果揭示了小分子在聚合物中的2种运动形式,一是小分子在聚合物空穴范围内震荡,二是小分子在聚合物空穴间的跳跃式迁移。从图2可以看出,噻吩分子先在空穴内震荡,当噻吩分子有足够的能量,或者当聚合物链运动产生了合适的路径后,发生空穴间迁移。通过这种方式,噻吩分子在聚合物中进行传质扩散。分子扩散轨迹所展现的扩散方式与通常认为的分子在聚合物中跳跃扩散的形式相一致[7]。从图2还可以看出,噻吩分子在EC体系中跳跃次数最少,大部分在固定的空穴中震荡,可以认为在EC中分子更难以扩散。

图2 噻吩分子在3种聚合物中的扩散轨迹
2.2 小分子在聚合物中的扩散速率
在MD模拟中,分子扩散速率常采用Einstein方法计算,Einstein法将扩散速率(D)与分子的均方位移(MSD)相关联,如式(1)[5]所示。
(1)
式(1)中,(ri(t)-ri(0))2为i组分的均方位移,ri(t)为t时间分子i的坐标,ri(0)为分子i的初始坐标。该方程假设分子在聚合物中自由运动,MSD与D呈线性关系,相应的扩散速率是均方位移对时间的变化率。通常,在分子动力学模拟的初始阶段会发生非正常扩散,分子扩散受限;在足够长的扩散时间后,扩散分子从非正常扩散转变为正常扩散。是否达到正常扩散可以通过(ri(t)-ri(0))2∝tn检验,n为时间t的指数;n<1表明在聚合物内小分子为非正常扩散,n=1表明在聚合物内小分子为正常扩散[10]。图3为噻吩在3种膜材料中MSD与t双对数曲线。可以看出,随着模拟时间增长,斜率n趋近于1,表明噻吩在3种聚合物膜材料中均由非正常扩散转变为正常扩散,因此2500 ps的模拟时间对笔者考察的膜材料体系来说足够长。通过式(1)求取汽油组分在3种聚合物膜材料中的扩散速率,结果示于图4。这3种聚合物渗透气化膜的脱硫性能列于表2。

图3 噻吩在不同聚合物体系中MSD与模拟时间(t)的对数曲线
从图4可以看出,在PEG和PDMS体系中,噻吩、正庚烷、环己烯、环己烷、甲苯分子的扩散速率依次降低,而在EC体系中扩散速率降低的顺序变为噻吩、甲苯、正庚烷、环己烯、环己烷。在EC、PEG和PDMS体系中,噻吩分子的扩散速率均大于其余4组分,表现出优先扩散性,且其在EC中扩散选择性最大,与表2所示EC、PEG和PDMS渗透气化膜的脱硫性能相符合[8]。同一扩散分子在EC体系中扩散速率最小,在PDMS体系中扩散速率最大,与图2观察分子扩散轨迹所得结论相一致。

图4 模拟汽油组分在不同聚合物体系中的扩散速率(D)

表2 实验所得3种聚合物膜材料的脱硫性能
1) Composed ofn-heptane, cyclohexene, cyclohexane, toluene and thiophene; 2) The sulfur enrichment factor, which represents the selectivity of sulfur removal.
2.3 影响小分子在聚合物体系中扩散的因素
2.3.1 扩散分子与聚合物相互作用能
小分子在聚合物体系中的扩散过程中,其与体系之间的非键相互作用具有很大的影响,可以通过扩散分子与体系的相互作用能ΔE来表征,如式(2)所示。
ΔE=(Emol+Esys)-Etol
(2)
式(2)中,Emol为扩散分子能量,Esys为去除扩散分子后混合体系的能量,Etol为总混合体系能量,单位均为kJ/mol。ΔE越大表明相互作用能越强,分子扩散离开接触面需要克服更高的能量势垒[6]。
本研究中,扩散分子与聚合物仅是物理混合,并无化学键作用,因此使用COMPASS力场通过公式(2)计算得到扩散分子与体系非键相互作用能ΔE,结果列于表3。从表3可见,在3种聚合物膜材料中,噻吩的ΔE均低于其余4种扩散分子的,噻吩扩散离开接触面需要的能量最低,最易于扩散,这与计算的扩散速率结果相符,表明扩散分子与聚合物相互作用是噻吩类硫化物在脱硫膜材料中优先扩散的决定因素,与Ling等[6]所得结论相一致。
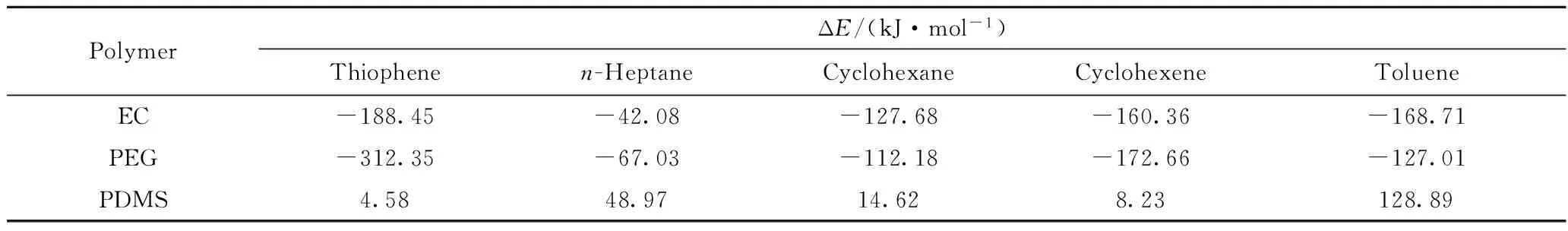
表3 扩散组分与聚合物的相互作用能(ΔE)
在3种聚合物膜材料中,噻吩、环己烷、环己烯和甲苯均体现出ΔE越大分子扩散速率越低的规律,而正庚烷在EC和PDMS中的ΔE最大,但其扩散速率却不是最低;就同一扩散分子在3种聚合物体系中的ΔE而言,与PDMS的ΔE最大,而其在PDMS体系中扩散速率却大于在EC和PEG体系中的(见图4),说明ΔE是影响扩散速率的重要因素,但不是单一决定因素。
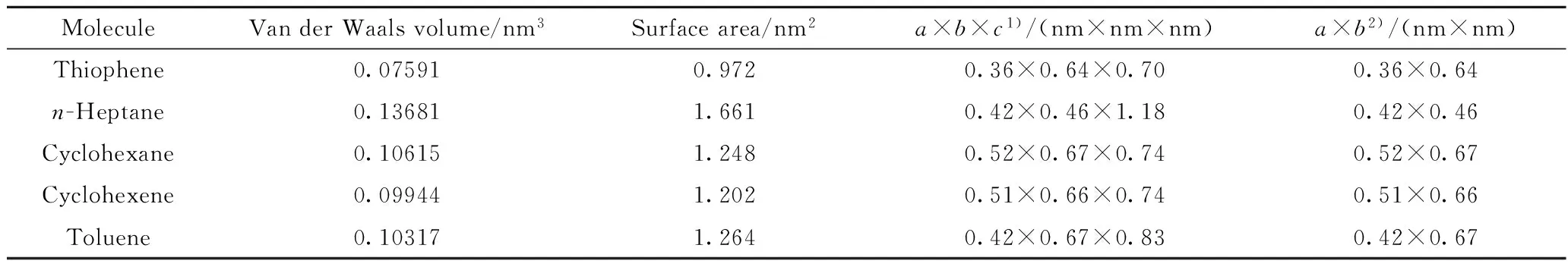
2.3.2 扩散分子尺寸
扩散分子的尺寸是影响分子扩散速率的重要因素。随着扩散分子尺寸的增大,其发生空穴间迁移的几率减小,传质的阻力增大[11]。利用Connoly Surface计算了扩散分子范德华体积和表面积,分子体积大小依次是正庚烷、环己烷、甲苯、环己烯、噻吩。然而分子体积仅能体现分子总体积的大小,不能描绘分子的空间形态和结构。为了更精确、全面地表达分子的形状和尺寸,使用Corey-Pauling-Koltun(CPK scale:1.0)模型模拟分子最低能量构象,将其作三维长方体模型处理。图5为正庚烷最低能量构象三视图。用分子在3个垂直方向上的长度a×b×c表示分子的三维尺寸,且a 由表4可以看出,虽然正庚烷的体积最大,但其最小截面尺寸明显小于其余4种分子,所以正庚烷沿着最小截面垂直方向扩散时阻力更小,需要的自由体积更小,更易于扩散[12]。虽然正庚烷扩散时需要克服的能量势垒要大于环己烷和环己烯,但其最小截面尺寸远小于环己烷和环己烯,扩散时有更多的空间和路径,因此表现出的扩散速率大于环己烷和环己烯。可以看出,扩散分子尺寸和分子与体系相互作用能2种因素相互协同,共同影响分子在聚合物中的扩散性。 图5 正庚烷最低能量构象三视图 表4 扩散分子尺寸 1) Molecular three-dimensional dimension; 2) Minimum cross-sectional dimension 2.3.3 聚合物自由体积 根据自由体积理论[13],聚合物总体积由占有体积和自由体积构成。聚合物自由体积孔穴的分布和大小对扩散分子在体系中的扩散行为具有重要影响[6]。普遍采用正电子湮没技术来测定材料的自由体积大小,然而实验方法经常存在较大误差,且无法精确描绘出自由体积的微观形貌。分子动力学方法采用特定原子作为探针来探测致密膜的自由体积,结果较为准确[5]。体系内被探测出的自由体积占总体积的比例称为自由体积分数(FFV),可由式(3)计算。 (3) 式(3)中,Vf和Vo分别为聚合物的自由体积和聚合物链的占据体积,nm3。 膜材料中体积较小的自由体积孔穴对膜的传递性质影响很小,能够容纳扩散分子的自由体积空穴对分子扩散有较大影响[14]。笔者利用噻吩为探测分子,用动力学直径为0.53 nm的硬球表示,考察不同膜材料容纳噻吩分子的自由体积分布及其所占比例。根据式(3)求得EC、PEG和PDMS自由体积分数分别为0.739%、0.217%和2.574%。聚合物自由体积空穴的模拟形貌示于图6,其中灰色和蓝色部分为自由体积空穴。可以看出,EC和PEG体系中自由体积空穴分散,而PDMS中自由体积空穴较为连续、紧密。聚合物的FFV越大,能为噻吩提供的扩散空间就越大, FFV从大到小的聚合物体系依次为PDMS、EC、PEG,而噻吩在这3种聚合物中的扩散速率大小却依次为PDMS、PEG、EC。虽然EC具有比PEG更多扩散空穴,但还要考虑扩散分子在聚合物中扩散方式,在EC中扩散分子主要在空穴内震荡,很难发生空穴间迁移。可见,不同膜材料自由体积大小并不是同一分子扩散速率大小的决定因素。 图6 3种聚合物自由体积空穴的模拟形貌 2.3.4 聚合物分子链运动性 聚合物分子链运动一定程度上决定了自由体积空穴间通道的形成。聚合物分子链运动越强,小分子越容易发生空穴间跃迁,表现出越高的扩散速率。聚合物分子链的运动性强弱也可以用分子链的自扩散速率(Ds)表示[15](见表1)。PDMS、PEG、EC 3种聚合物的Ds依次降低,与扩散组分在这3种材料中扩散速率大小顺序一致。虽然EC的自由体积分数比PEG大,但是EC分子链运动性远小于PEG,EC中自由体积之间很难形成有效通道,扩散分子在固定空穴中震荡,导致分子在EC膜中扩散速率小于在PEG中的。自由体积和聚合物分子链运动的重要性也从侧面验证了分子在空穴间跳跃扩散的扩散方式。 EC分子链的运动性远差于PEG和PDMS的,为分析造成这种差异的原因,笔者模拟计算了这3种材料的氢键作用力和玻璃化转变温度(见表1)。通常情况下,氢键是最强的分子间作用力,虽然其小于化学键,但其对材料的微观形貌起重要作用[7];玻璃化转变温度(Tg)体现聚合物分子链的软硬程度,是对比聚合物分子链运动性的重要指标[7],玻璃化转变温度越低,聚合物分子链运动性越强。计算结果表明,EC膜体系中氢键作用力远大于PEG和PDMS的,EC分子间大量氢键制约其分子链运动;对比聚合物玻璃化温度与其分子链运动性发现,在体系模拟温度(330.15 K)未达到EC的玻璃化转变温度时,EC分子链几乎不运动,导致很难形成有效的分子扩散路径。 (1) 采用分子动力学方法和COMPASS力场模拟了噻吩、正庚烷、环己烷、环己烯和甲苯分子在EC、PEG和PDMS膜脱硫材料中的扩散方式,并计算了分子的扩散速率。结果表明,噻吩在EC、PEG和PDMS膜材料中均表现出优先扩散性,与这3种材料表现出的脱硫性能相符。 (2) 分析了扩散分子与聚合物的相互作用能、扩散分子尺寸、聚合物自由体积分数和聚合物分子链运动性对分子扩散的影响,考察了扩散分子在聚合物中微观扩散机理。结果表明,扩散分子与聚合物相互作用是噻吩类硫化物在脱硫膜材料中优先扩散的决定因素,且扩散分子与体系的相互作用能越低、扩散分子尺寸越小、聚合物自由体积分数越大、聚合物分子链运动性越强,越有利于扩散分子在聚合物中的扩散。各因素相互协同共同影响扩散分子在聚合物中的扩散速率。 [1] 孔瑛, 卢福伟, 吕宏凌, 等. 渗透汽化膜法汽油脱硫技术研究进展[J].膜科学与技术, 2011, 31(3): 162-171.(KONG Ying, LU Fuwei, LU Hongling, et al. The research progress of gasoline desulfurization by pervaporation[J].Membrane Science and Technology, 2011, 31(3): 162-171.) [2] 林立刚, 张玉忠. 膜技术在汽油脱硫中应用的新进展[J].石油学报(石油加工), 2010, 26(3): 476-485.(LIN Ligang, ZHANG Yuzhong. Recent advances in sulfur removal from gasoline by membrane process[J].Acta Petrolei Sinica (Petroleum Processing Section), 2010, 26(3): 476-485.) [3] 卢福伟, 孔瑛, 吕宏凌, 等. 分子动力学模拟噻吩/正庚烷在PEG膜中溶解-扩散行为[J].高分子材料科学与工程, 2012, 28(3): 114-117.(LU Fuwei, KONG Ying, LÜ Hongling, et al. Molecular dynamics simulation studies on sorption and diffusion properties of thiophene/heptane in peg membranes[J].Polymer Materials Science and Engineering, 2012, 28(3): 114-117.) [4] 王俊, 朱宇, 陆小华. 分子模拟在聚合物膜研究中的应用[J].现代化工, 2003, 23(10): 59-62.(WANG Jun, ZHU Yu, LU Xiaohua. Application of molecular simulation in polymeric membrane science[J].Modern Chemical Industry, 2003, 23(10): 59-62.) [5] LI Ben, PAN Fusheng, FANG Zhiping, et al. Molecular dynamics simulation of diffusion behavior of benzene/water in PDMS-Calix[4]arene hybrid pervaporation Membranes[J].Ind Eng Chem Res, 2008, 47(13): 4440-4447. [6] LING Changjian, LIANG Xiaoyi, FAN Feichao, et al. Diffusion behavior of the model diesel components in different polymer membranes by molecular dynamic simulation[J].Chemical Engineering Science, 2012, 84: 292-302. [7] AMANI M, AMJAD-IRANAGH S, GOLZAR K, et al. Study of nanostructure characterizations and gas separation properties of poly(urethane-urea)s membranes by molecular dynamics simulation[J].Journal of membrane Science, 2014, 462: 28-41. [8] RYCHLEWSKA K, KONIECZNY K, BODZEK M. Pervaporative desulfurization of gasoline separation of thiophene/n-heptane mixture[J].Archives of Environmental Protection, 2015, 41(2): 3-11. [9] 林立刚. 渗透汽化膜分离法脱除汽油中有机硫化物的研究[D].青岛: 中国石油大学(华东), 2008. [10] GOLZAR K, AMJAD-IRANAGH S, AMANI M, et al. Molecular simulation study of penetrant gas transport properties into the pure and nanosized silica particles filled polysulfone membranes[J].Journal of Membrane Science, 2014, 451: 117-134. [11] 卢福伟. 共聚高分子膜材料微相结构与汽油脱硫性能关系研究 [D].青岛: 中国石油大学(华东), 2011. [12] 袁帅, 龙军, 田辉平, 等. 烃分子尺寸及其与扩散能垒关系的初步研究[J].石油学报(石油加工), 2011, 27(3): 376-380.(YUAN Shuai, LONG Jun, TIAN Huiping, et al. Molecular dimensions of hydrocarbons and a primary study on its relationship to diffusion energy barriers in zeolites[J].Acta Petrolei Sinica (Petroleum Processing Section), 2011, 27(3): 376-380.) [13] LIPATOV Y. The iso-free-volume state and glass transitions in amorphous polymers[J].Advances in Polymer Science, 1978, 26: 63-104. [14] 潘福生. 超薄复合膜的理性设计、结构调控及脱湿性能研究[D].天津: 天津大学, 2009. [15] PAN Fusheng, MA Jing, CUI Li, et al. Water vapor/propylene sorption and diffusion behavior in PVA-P(AA-AMPS) blend membranes by GCMC and MD simulation[J].Chemical Engineering Science, 2009, 64(24): 5192-5197. Diffusion Behavior of the Model Gasoline Components inDifferent Polymer Membranes Studied by Molecular Dynamic Simulation HOU Yingfei1,2, JIANG Chi1, LIU Min1, LIU Yibin1, LI Peng1, NIU Qingshan1 (1.StateKeyLaboratoryofHeavyOilProcessing,ChinaUniversityofPetroleum,Qingdao266580,China;2.DepartmentofPetroleumandChemicalEngineering,XinjiangVocationalandTechnicalCollegeofApplication,Kuitun833200,China) To explore the main factor in desulfurization performance of pervaporation membranes, molecular dynamic (MD) simulation was used to investigate the diffusion behavior of model gasoline components (thiophene,n-heptane, cyclohexane, cyclohexene, and toluene) in three different membranes of ethyl cellulose (EC), polyethylene glycol (PEG), and polydimethylsiloxane (PDMS). Trajectories of molecules diffusing were drawn by obtaining the barycentric coordinates of gasoline components in three polymer membranes. Diffusion coefficients of five gasoline components in different membranes were calculated, and the micro diffusion mechanism of diffusion molecule inside polymer membrane was investigated by analyzing the interaction energy between diffusion molecule and polymer membrane, the size of diffusion molecule, fractional free volume (FFV) of polymer membrane and the mobility of polymer chains. It is obviously discovered that the diffusion coefficients of thiophene were considerably greater than other components in three different membranes, which agreed well with the experimental results. Overall, the diffusion coefficients were attributed to the combined effect of the four factors. molecular dynamic; molecular diffusion; gasoline desulfurization; membrane separation 2015-12-29 国家自然科学基金项目(51173203)、中央高校基本科研业务费专项资金项目(15CX05014A)、山东省自然科学基金项目(ZR2012BL15)资助 侯影飞,男,副教授,博士,从事石油与天然气加工、膜分离、油田化学品等研究;Tel: 0532-86984702;E-mail: houyf@upc.edu.cn 1001-8719(2016)06-1121-07 TQ028.8 A 10.3969/j.issn.1001-8719.2016.06.006

3 结 论
猜你喜欢
车用发动机(2021年5期)2021-10-31
赤峰学院学报·自然科学版(2018年7期)2018-08-11
小型内燃机与车辆技术(2016年4期)2016-10-21
浙江大学学报(工学版)(2016年11期)2016-06-05
当代化工研究(2016年1期)2016-03-16
合成化学(2015年9期)2016-01-17
合成化学(2015年10期)2016-01-17
化工进展(2015年6期)2015-11-13
应用化工(2014年9期)2014-08-10
郑州大学学报(工学版)(2014年6期)2014-03-01
