
氨基化修饰介孔Fe3O4@SiO2@mSiO2磁性核壳复合微球的可控制备及吸附性能
2016-12-15 07:42薛娟琴徐尚元朱倩文强亮生马晶
无机化学学报 2016年9期
薛娟琴 徐尚元 朱倩文 强亮生 马晶
氨基化修饰介孔Fe3O4@SiO2@mSiO2磁性核壳复合微球的可控制备及吸附性能
薛娟琴*,1徐尚元1朱倩文1强亮生2马晶*,1
(1西安建筑科技大学化学工程与工艺系,西安710055)
(2哈尔滨工业大学化工与化学学院,哈尔滨150001)
以水热法制备的高磁饱和强度Fe3O4纳米颗粒为核,正硅酸乙酯(TEOS)为前驱体,采用改进的Stöber法,制备介孔SiO2包覆Fe3O4磁性核壳复合微球。利用XRD、SEM、TEM、N2吸附-脱附、FTIR和VSM对制备样品的物相结构、形貌和磁性能进行了测试表征。研究结果表明,制备的复合材料呈球形,粒径分布均一,材料的比表面积和磁饱和强度分别为413 m2·g-1和68.93 emu·g-1。研究了TEOS的添加量对复合微球形貌的影响,随着TEOS添加量的增加,SiO2壳层增厚,复合粒子形貌均匀,饱和磁化强度有所下降,仍具有良好的超顺磁性。在此基础上,通过接枝法在复合微球的表面接枝-NH2,制备了一种新型磁性纳米吸附剂(Fe3O4@SiO2@mSiO2-NH2),进而研究了其对水中重金属离子Cr(Ⅳ)的吸附性能。通过动力学拟合,Fe3O4@SiO2@mSiO2-NH2对Cr(Ⅳ)的吸附过程是准二级动力学模型占主导地位,探究了该材料对Cr(Ⅳ)的吸附过程和吸附机理。结果表明,其吸附机理及吸附容量与Cr(Ⅳ)的离子形态及-NH2有关,并通过吸附剂与吸附质之间的电子共用或静电吸附实现。
核壳结构;复合磁性微球;介孔二氧化硅;吸附性能
随着纳米材料的快速发展,纳米磁性复合微球作为一类重要的功能材料由于具有许多优异的性能,已成为相关领域的研究热点[1]。纳米Fe3O4作为典型的磁性纳米粒子,以其独特磁学性质、较好生物相容性和毒副作用小等特点而备受关注,并常作为磁核来制备磁性复合材料,应用于众多领域。但由于磁性纳米粒子自身的磁偶极矩作用,使其易团聚和氧化,因此有必要对磁性纳米粒子进行表面修饰[2-5]。近年来有越来越多的研究关注于SiO2材料,由于其具有很好的相容性以及表面充足的SiOH基团易于改性,使其成为了一种理想的修饰材料[6-7]。而SiO2包覆Fe3O4复合材料(Fe3O4@SiO2)具有几个突出的特点:(1)SiO2稳定性高,可提高其生物相容性、亲水性;(2)纳米粒子表面包覆SiO2可防止Fe3O4氧化,提高粒子的稳定性,阻止粒子发生团聚;(3)材料表面富含大量的羟基官能团,利于进一步功能化。
由于Fe3O4@SiO2具有超顺磁性,能够在外加磁场的作用下实现快速的分离。因此,其在重金属离子吸附处理方面的应用研究引起了研究者的兴趣。但磁性颗粒的引入导致了比表面积和孔容减小[8]。如何设计一种合理的复合材料,集Fe3O4、SiO2的优点于一体,利用两者之间的有效协同作用,能够对重金属离子进行快速吸附,是目前亟待解决的技术课题。
一般情况下,Fe3O4@SiO2磁性复合材料的合成是在醇水体系中通过正硅酸乙酯缩合的溶胶凝胶方法(Stöber法),但是由于常规的磁性液体基本都是油溶性的,使得Stöber法难以在磁性材料外层包覆二氧化硅,且制备的纳米粒子粒径不均、难以控制。为了解决这一问题,有研究者采用超声辅助方法制备[9],但该工艺复杂难以精确控制,且同相成核严重;或采用反相微乳液法制备[10],使互不相溶的物质能溶于同一个体系,但此法容易引入杂质。为此,Shi等采用赤铁矿为磁源,在其外围包覆一层二氧化硅之后,通过还原反应得到了颗粒尺寸分布均匀的Fe3O4@SiO2复合微球[11]。Fu等基于Fe3O4纳米粒子的亲水性和十二烷基硫酸钠的乳化剂性质,采用蒸发法制备了十二烷基硫酸钠修饰的Fe3O4纳米粒子[12]。Wang等以阳离子型高分子材料聚乙烯吡咯烷酮为分散剂,通过与Fe3O4纳米粒子发生静电作用形成稳定的水溶性磁流体,制得粒径均一、核内Fe3O4分布均匀的核壳型Fe3O4@SiO2磁性复合纳米粒子[13]。
本文通过改进的Stöber法,制备了介孔SiO2包覆Fe3O4磁性核壳复合微球。这种新型结构的磁性材料层和介孔材料层的制备过程相对独立,内部SiO2层的致密结构起到稳定和保护Fe3O4纳米颗粒的作用;外部SiO2层的多孔结构带来丰富的孔隙率和大的比表面积,则赋予复合材料良好的吸附性能。在此基础上,通过接枝法在磁性核壳复合微球的表面接枝-NH2,制备了一种新型磁性纳米吸附剂(Fe3O4@SiO2@mSiO2-NH2),进而研究了其对水中重金属离子Cr(Ⅳ)的吸附性能。由于Fe3O4纳米结构具有超顺磁性能而有利于颗粒的回收再利用,同时这种新型结构的复合微球具有孔径可调、尺寸均一及无生理毒性的特点,因此在生物催化、吸附及分离等方面有较大的应用价值[14-15]。
1 实验部分
1.1 主要试剂
三氯化铁(FeCl3·6H2O)、柠檬酸三钠(C6H5Na3O7· 2H2O)、无水乙酸钠(CH3COONa)、正硅酸四乙酯((C2H5O)4Si,TEOS)均为分析纯,购于天津市福晨化学试剂厂;乙二醇(HOCH2CH2OH)、无水乙醇(C2H5OH)、十六烷基三甲基溴化铵(C19H42BrN,CTAB)、聚乙二醇20000均为分析纯,购于天津市科密欧化学试剂有限公司;3-氨丙基三甲氧基硅烷(C16H17NO3Si),购于上海阿拉丁生化有限公司;去离子水自制。
1.2 材料合成
1.2.1 Fe3O4的制备
参照文献方法制备Fe3O4[16],将1.35 g FeCl3· 6H2O和40 mL乙二醇混合均匀,加入3.6 g无水乙酸钠和1 g聚乙二醇,超声和磁力搅拌交替进行30min,使混合物充分溶解后,将混合液转入以聚四氟乙烯为内衬的不锈钢反应釜中,200℃晶化8 h,产物自然冷却后,先后用去离子水和无水乙醇清洗多次,干燥得Fe3O4磁性纳米粒子。
1.2.2 介孔Fe3O4@SiO2@mSiO2磁性核壳复合微球
采用改进的Stöber法合成介孔Fe3O4@SiO2@ mSiO2磁性核壳复合微球[17],制备流程如图1所示。具体过程如下:
称取0.15 g Fe3O4纳米粒子,分散到50 mL去离子水中,分别用10 mL 2 mol·L-1的HCl和100 mL 0.5 mol·L-1的柠檬酸三钠溶液对其进行改性。柠檬酸钠吸附在Fe3O4纳米粒子表面,改变其表面电位,利于Fe3O4纳米粒子克服磁偶极矩。
将改性后的Fe3O4超声分散于含有氨水(25%, w/w)的乙醇水溶液中,在机械搅拌的同时缓慢滴加TEOS,滴加完毕后继续搅拌24 h,得到单层SiO2包覆Fe3O4复合材料(Fe3O4@SiO2)。所得产物用水和乙醇清洗数次,直至溶液呈中性。将所得Fe3O4@SiO2分散于60 mL乙醇、80 mL去离子水和4 mL氨水(25%,w/w)的混合溶液中,再加入0.6 g模板剂CTAB,超声分散30 min后,将混合溶液转移至三口烧瓶中搅拌2 h。之后缓慢滴加TEOS,继续搅拌24 h。所得产物用水和无水乙醇清洗数次,在60℃真空干燥12 h。去除模板剂CTAB采用离子交换法,将获得的产物分散于硝酸铵-乙醇溶液中,在60℃水浴中搅拌2 h,重复数次。样品命名为Fe3O4@SiO2@mSiO2(x),x为加入的TEOS体积量(mL)。
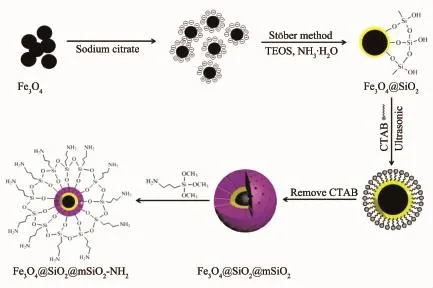
图1 氨基化修饰介孔Fe3O4@SiO2@mSiO2磁性核壳复合微球制备流程Fig.1 Preparation diagram of Fe3O4@SiO2@mSiO2
1.2.3 氨基化修饰介孔Fe3O4@SiO2@mSiO2磁性核壳复合微球
称取已干燥的介孔Fe3O4@SiO2@mSiO2复合微球,加入250 mL的三口圆底烧瓶中,再加入一定量的无水乙醇,超声分离20~25 min。按照复合微球与硅烷试剂3-氨丙基-三甲氧基硅烷的质量体积比为1∶1.05~1∶1.2加入硅烷试剂,加入后在80~100℃下回流9~14 h并搅拌,其目的是使硅烷试剂吸附到SiO2的介孔孔道内,并发生水解反应。依次采用磁分离收集、丙酮回流洗涤12~48 h、0.1~2 mol·L-1酸溶液洗涤、去离子水洗涤的工艺,去除未反应或未吸附到介孔孔道内的硅烷试剂,最终经真空干燥得到氨基修饰介孔Fe3O4@SiO2@mSiO2,样品命名为Fe3O4@SiO2@mSiO2-NH2。
1.3 材料表征
采用日本岛津的XRD-6000型X射线衍射仪分析样品的结构及Fe3O4晶型。工作管电压40 kV,管电流50 mA,辐射源为Cu靶Kα(λ=0.154 18 nm),扫描范围20°~80°。小角XRD(low-angle XRD)是判断材料是否具有介孔孔道的最有效方法之一,利用Bruker D8 X-射线粉末衍射仪对合成的样品进行介孔结构表征。辐射源为Cu靶Kα(λ=0.154 18 nm),扫描范围0.5°~4.5°,步长0.02°,扫描速度0.5°· min-1,管电流50 mA,管电压40 kV。利用康塔公司Autosorb-1比表面和孔隙度分析仪测试比表面积。测试前样品在真空条件下于150℃脱气8 h。由BET方程计算样品的比表面积,用BJH方法计算孔径分布曲线。利用日本电子日立S4800型场发射扫描电子显微镜研究样品微观结构和晶粒形貌,工作电压15 kV。采用日立H-800型透射电子显微镜(加速电压200 kV)观察样品的微观形貌和核壳结构;采用KBr固体压片法,在Nicolet Magna 550型红外光谱仪上测定样品红外(FTIR)光谱,分析样品的组分和所带的官能团。采用振动样品磁强计(美国Lakeshore 7307VSM)测量样品的磁滞回线。
1.4 吸附性能测试
1.4.1 吸附实验
为了研究材料的吸附性能,选用重金属离子Cr(Ⅳ)作为模型吸附质,采用静态吸附实验考察材料对不同浓度Cr(Ⅳ)吸附性能。首先量取100 mL特定浓度的Cr(Ⅳ)水溶液,Cr(Ⅳ)的浓度分别选择10.0、20.0、30.0 mg·L-1;然后在搅拌条件下加入0.05 g吸附剂进行吸附,一定时间后,通过外加磁场将吸附剂从溶液中分离开来;最后取上清液用于测定吸附后溶液中的Cr(Ⅳ)浓度。
1.4.2 Cr(Ⅳ)的测定
水样中的Cr(Ⅳ)测定采用二苯碳酰二肼分光光度法[18]。
首先称取0.282 9 g K2Cr2O7溶于去离子水中,稀释至1 000 mL,摇匀备用。该溶液称为Cr(Ⅳ)标准储备液,Cr(Ⅳ)浓度为100 mg·L-1。然后吸取5 mL Cr(Ⅳ)标准储备液,用去离子水稀释至500 mL,摇匀,得到Cr(Ⅳ)浓度为1 mg·L-1的标准溶液。
向一系列50 mL的比色管中分别加入0、0.20、0.50、1.00、2.00、4.00、6.00、8.00和10.00 mL的Cr(Ⅳ)浓度为1 mg·mL-1的铬离子标准溶液,用水稀释至标线,加入1∶1硫酸溶液0.5 mL和1∶1磷酸溶液0.5 mL,摇匀,加入2 mL显色剂(二苯碳酰二肼的丙酮溶液),摇匀,静置5~10 min,通过紫外可见分光光度计(λ=540 nm)测定吸光度。作空白校正后,以540 nm处吸光度为纵坐标,Cr(Ⅳ)的含量(mg)为横坐标,绘制标准曲线,如图2所示。

图2 Cr(Ⅳ)标准曲线Fig.2 Chromium cation standard curve
取1 mL上清液,以水为参比,置于50 mL比色管中,用水稀释到标线,使用上述方法测定样品540 nm处的吸光度,经过空白校正以后,从标准曲线上查得Cr(Ⅳ)含量。吸附百分比可通过式(1)计算:

式中,α为吸附百分比,c0和c分别为样品溶液中Cr(Ⅳ)的初始浓度和吸附平衡后溶液中Cr(Ⅳ)的浓度(m g·L-1)。

图3 Fe3O4@SiO2、Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2的广角XRD衍射图(a)和小角XRD衍射图(b)Fig.3 Wide-angle XRD patterns(a)and low-angle XRD patterns(b)of Fe3O4@SiO2,Fe3O4@SiO2@mSiO2and Fe3O4@SiO2@mSiO2-NH2
2 结果与讨论
2.1 XRD分析
图3(a)为Fe3O4纳米颗粒、Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2磁性核壳复合微球的广角XRD衍射图。由图可见:所合成的复合材料,均在30.02°、35.50°、43.1°、53.4°、57.12°和62.8°处出现了尖锐的特征衍射峰。通过与Fe3O4(PDF No.19-0629)衍射峰位对比可知,这些衍射峰分别对应(220)、(311)、(400)、(422)、(511)和(440)晶面的特征衍射峰。而Fe3O4@SiO2@mSiO2在2θ=22°~26°范围内具有宽吸收峰,该吸收峰为SiO2的无定形结构所致,这表明SiO2在包覆Fe3O4过程中,Fe3O4纳米颗粒的晶型结构并未发生改变。随着TEOS量的增加,30.08°所对应的晶面(311)的特征峰强度逐渐减弱,说明Fe3O4包覆的SiO2壳层厚度逐渐增加。同时,氨基的引入并没有改变Fe3O4@SiO2@mSiO2复合材料中Fe3O4的晶型及核壳结构。
图3(b)为Fe3O4@SiO2和磁性核壳复合微球的小角XRD衍射图。Fe3O4@SiO2@mSiO2-NH2在0.9°附近出现了较强的(100)衍射峰,说明其生成了短程有序的介孔结构。氨基的引入,样品仍在(100)出现较明显的衍射峰,证明引入NH2并未改变材料的介孔结构,但衍射强度有所降低,表明其有序度有所降低。
2.2 N2吸附-脱附分析
N2吸附-脱附等温曲线是表征介孔材料的重要手段。图4是磁性核壳复合微球的N2吸附-脱附等温曲线。测试结果表明,Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2吸脱附曲线属于第Ⅳ类等温线,并出现明显的滞后环,这表明材料具有介孔结构[19]。在相对压力略低的较窄区间(p/p0=0.40~0.75)内,氮吸附量随N2分子在介孔孔道内发生毛细凝聚而急剧增加,表明孔径较小且尺寸分布较集中,符合典型介孔分子筛的特征。
如图4内插图所示,Fe3O4@SiO2的N2吸附-脱附曲线则可归为第Ⅱ类等温线。在p/p0=0.9附近出现微小的滞后环,可归为材料的非致密结构,这主要由于第一层SiO2包覆时,内部形成的空心腔所致,说明材料存在微孔结构[20]。
表1为Fe3O4@SiO2、Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2的孔结构参数。由表可知,Fe3O4@ SiO2@mSiO2的比表面积和总孔容分别为416 m2·g-1和0.23 cm3·g-1,远大于Fe3O4纳米颗粒(8 m2·g-1和0.019 cm3·g-1)和Fe3O4@SiO2的比表面积和总孔容(256 m2·g-1和0.12 cm3·g-1),且与Fe3O4@SiO2@mSiO2-NH2相近。Fe3O4@SiO2@mSiO2的孔径为3.12 nm,也大于Fe3O4@SiO2的相应值(2.06 nm)。

图4 Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2的N2吸附-脱附等温线Fig.4 N2adsorption-desorption isotherms of Fe3O4@SiO2@mSiO2and Fe3O4@SiO2@mSiO2-NH2

表1 磁性核壳复合微球的孔结构参数Table 1 Pore structure parameter of composite magnetic microsphere
2.3 红外光谱(FTIR)分析
图5为Fe3O4纳米颗粒、Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2磁性核壳复合微球的红外光谱。其中,1 100 cm-1处强而宽的吸收峰为Si-O-Si反对称伸缩振动吸收峰,797 cm-1处的吸收峰则为Si-O-Si的对称伸缩振动峰和弯曲振动峰,3 434 cm-1处的吸收峰对应磁性纳米颗粒表面吸附水的伸缩振动,1 635 cm-1处的吸收峰对应-OH的弯曲振动[21]。此外,在Fe3O4的谱线上,594 cm-1处的宽吸收带对应于Fe3O4的Fe-O的伸缩振动,当Fe3O4表面包覆了SiO2后Fe-O特征峰从594 cm-1移到585 cm-1处,发生了“红移”[22]。Fe3O4@SiO2@mSiO2-NH2的谱线上,在1 546 cm-1处出现的峰为N-H的特征吸收峰,说明-NH2已成功修饰在Fe3O4@SiO2@mSiO2磁性复合微球的表面[23]。
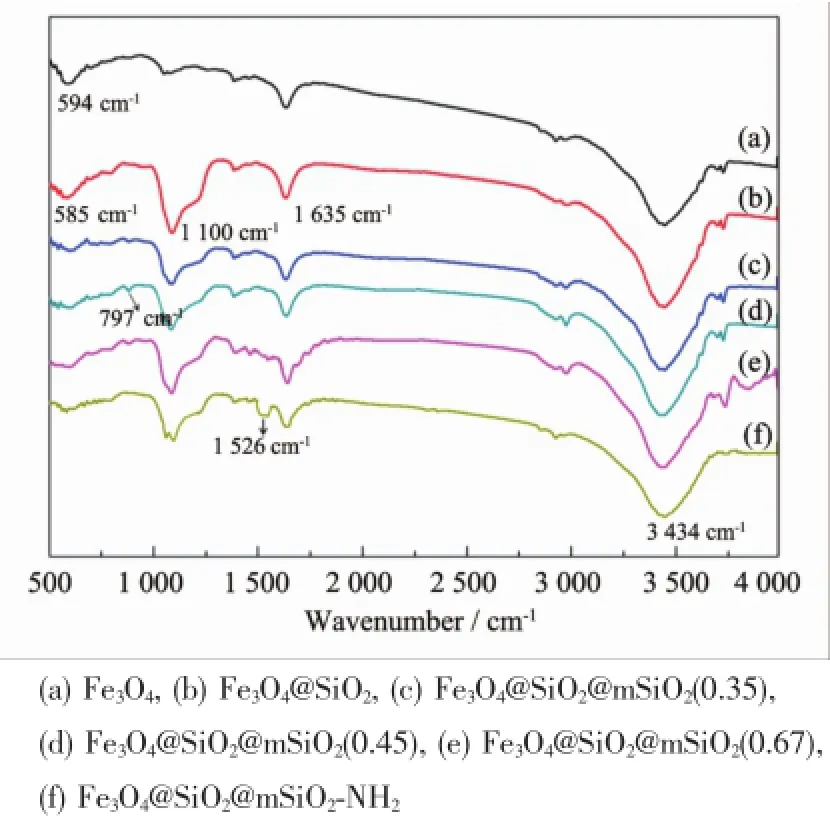
图5 磁性核壳复合微球的红外光谱图Fig.5 FTIR spectra of composite magnetic microsphere
2.4 样品的形貌分析
图6(a)~(c)分别为所得磁性Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@mSiO2的SEM照片。由图6(a)可以看出,制备的Fe3O4纳米颗粒为均匀的球形,但表面粗糙,粒径分布均一,并且分散性良好。在包覆SiO2后,Fe3O4的表面变得光滑,且粒径分布均一,分散性并未发生明显变化。
采用TEM对Fe3O4纳米颗粒、Fe3O4@SiO2和Fe3O4@SiO2@mSiO2磁性核壳复合微球的外观形貌、粒径进行了表征。由图6(d)可见,Fe3O4纳米颗粒呈球形,且大小均匀、分散性较好,平均粒径在100nm左右。由图6(e)~(h)可清晰地观察到,所有的粒子中黑色的Fe3O4核被灰色的SiO2壳层所包覆,且核壳微球表面光滑。包埋于SiO2内部的Fe3O4纳米粒子清晰可见,复合纳米粒子呈现明显的核壳结构。且随着TEOS添加量增加,核壳微球的粒径变大,SiO2壳层厚度增加,表面形貌更均匀,这是由于进入水核内的TEOS水解产物的质量浓度变大,导致Fe3O4粒子表面的SiO2壳层厚度增加;且随壳层厚度的增加,Fe3O4@SiO2@mSiO2复合粒子形貌受Fe3O4粒子形貌的影响变小,使其更趋向于规则的球形。此外,TEOS添加量的增加,还出现了SiO2包覆多个Fe3O4磁性纳米颗粒的现象,这可能是由于TEOS浓度较大,导致溶液离子强度增大,粒子表面电位下降,Fe3O4颗粒在被SiO2包覆前发生了一定程度的失稳和团聚所致[24]。结果显示,当TEOS的用量分别为0.35、0.45、0.67 mL时,SiO2壳层厚度分别为22.0、28.4和32.8 nm。同时氨基引入之后,Fe3O4@SiO2@mSiO2可以观察到规整的孔道结构(图6(h)内插图所示),这与小角XRD的分析结果相吻合。

图6 加入不同体积量TEOS得到磁性核壳复合微球的形貌分析Fig.6 SEM and TEM images of composite microspheres with various volumes of TEOS
2.5 样品的磁性能分析
图7为Fe3O4纳米颗粒、Fe3O4@SiO2和Fe3O4@ SiO2@mSiO2磁性核壳复合微球的磁滞回线。由图7可知,Fe3O4纳米颗粒的饱和磁化强度为78.93emu·g-1,且具有良好的超顺磁性。在SiO2包覆后,由于Fe3O4@SiO2、Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@ mSiO2-NH2核壳微球中Fe3O4相对含量减少,使得复合粒子的饱和磁化强度有所下降。随着TEOS添加量的增加,Fe3O4@SiO2@mSiO2复合粒子饱和磁化强度也呈现出下降趋势,这是由于SiO2壳层逐渐增厚,导致Fe3O4的相对含量降低的缘故。包覆后,复合材料仍具有较好的超顺磁性。因此,可通过调节TEOS用量获得不同磁性能的复合纳米粒子。

图7 磁性核壳复合微球的磁滞回线Fig.7 Magnetic hysteresis loops of composite microspheres
Fe3O4@SiO2@mSiO2表面经过氨基功能化改性降低了磁性,但仍然完全可以在外磁场的作用下与溶液迅速分离。当在外加磁场作用下时,纳米粒子在1 min内便可全部从溶剂中分离,全部吸附在试剂瓶侧壁上,表明制备的Fe3O4@SiO2@mSiO2-NH2磁性核壳复合微球具有快速的磁响应性能。
2.6 样品的吸附性能
2.6.1 吸附性能对比
图8为Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO -NH2对不同浓度Cr(Ⅳ)溶液的吸附曲线。Fe3O4@SiO2@mSiO2-NH2的饱和吸附量明显大于Fe3O4@SiO2@mSiO2,这是因为功能化-NH2易与Cr(Ⅳ)形成配位化合物,形成特异性吸附。同时,带正电荷的-NH2引入,可与溶液中的带负电荷CrO42-通过静电作用,增强核壳微球的吸附能力。故Fe3O4@SiO2@mSiO2-NH2的饱和吸附量较大。虽然Fe3O4@SiO2@mSiO2具有相对大的比表面积和丰富的孔结构,但其对Cr(Ⅳ)的吸附能力较小,仅为3.4 mg·g-1。这主要归因于Fe3O4@SiO2@mSiO2介孔结构对Cr(Ⅳ)的非特异性结合,导致其吸附量较小。对于吸附重金属离子而言,除了需要发达的介孔结构外,合适的官能团修饰也是提高重金属离子吸附性能的关键。

图8 Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2对不同浓度Cr(Ⅵ)溶液的吸附曲线Fig.8 Adsorption-time curves of Fe3O4@SiO2@mSiO2and Fe3O4@SiO2@mSiO2-NH2
当同样量的吸附载体加入到不同初浓度的Cr(Ⅳ)溶液(10.0、20.0、30.0 mg·L-1)时,所得到的平衡浓度不同,吸附剂对其各自的吸附量也不同。初浓度对载体的吸附能力也是一个很大的影响因素。当溶液里含Cr(Ⅳ)的浓度很低时,载体所表现出的吸附能力较弱。随着溶液里Cr(Ⅳ)浓度的增加,载体所吸附的Cr(Ⅳ)量也增加,初浓度增至30.0 mg·L-1时,吸附量几乎不再变化,此时吸附基本达到饱和。
2.6.2 吸附动力学研究
为了进一步探讨磁性复合微球对Cr(Ⅳ)的吸附动力学特性,选用准一级动力学方程(式(2))和准二级动力学方程(式(3))对吸附动力学数据进行拟合[25-26]。

式中,Qt为t时刻的吸附量(mg·g-1);Qe为平衡吸附量(mg·g-1);k1为一级吸附速率常数(min-1),k2为二级吸附速率常数(g·mg-1·min-1)。
图9为Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2吸附10.0 mg·L-1Cr(Ⅳ)溶液动力学拟合曲线。可以发现,Fe3O4@SiO2@mSiO2-NH2在吸附Cr(Ⅳ)过程中,按准二级动力学模型拟合得到的k2和R2分别为8.56×10-3g·mg-1·min-1和0.978,远远大于准一级动力学模型所得R2(0.92),这表明准二级动力学方程比准一级动力学方程能更好地描述Fe3O4@ SiO2@mSiO2-NH2复合微球在室温下对Cr(Ⅳ)的吸附。而Fe3O4@SiO2@mSiO2对Cr(Ⅳ)的吸附则更符合准一级动力学方程。
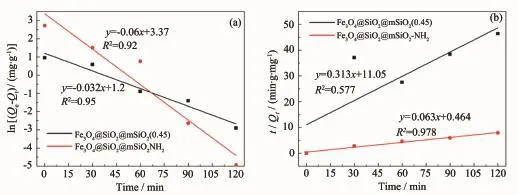
图9 Fe3O4@SiO2@mSiO2和Fe3O4@SiO2@mSiO2-NH2的动力学模型:(a)准一级动力学模型,(b)准二级动力学模型Fig.9 Kinetic model curves of Fe3O4@SiO2@mSiO2and Fe3O4@SiO2@mSiO2-NH2:(a)pseudo-first order kinetics; (b)pseudo-second order kinetics
准一级动力学模型说明吸附过程是由扩散步骤控制,属于物理吸附。而准二级动力学模型则假定吸附过程是由化学吸附控制的,是通过吸附剂与吸附质之间的电子共用或电子转移来实现的,为化学吸附过程。通过比较准一级动力学模型和准二级动力学模型的拟合曲线的相关系数,可得结论:所合成的Fe3O4@SiO2@mSiO2-NH2的吸附过程中准二级动力学模型占主导地位,且整个过程是通过吸附剂与吸附质之间的电子共用或静电吸附来实现的,是化学吸附过程;而Fe3O4@SiO2@mSiO2的吸附过程则只是通过介孔结构实现简单的物理吸附。
3 结论
以高磁饱和强度Fe3O4纳米颗粒为核,正硅酸乙酯(TEOS)为前驱体,采用改进的Stöber法,制备了介孔结构SiO2包覆Fe3O4磁性核壳复合微球。在此基础上,通过接枝法在复合微球的表面接枝-NH2,制备了一种新型磁性纳米吸附剂(Fe3O4@SiO2@mSiO2-NH2)。利用XRD、SEM、TEM、VSM、FTIR和N2吸附-脱附对制备样品的物相结构、形貌和磁性能进行了测试表征。制备的磁性核壳复合微球呈球形,包覆介孔SiO2样品的磁饱和强度有所下降,且随着加入TEOS体积量的增加,SiO2壳层增厚。复合磁性核壳微球的比表面积和孔容分别为413 m2·g-1和0.21 cm3·g-1。在对Cr(Ⅳ)的吸附过程中,Fe3O4@SiO2@ mSiO2-NH2的吸附过程中准二级动力学模型占主导地位,其吸附机理及吸附容量与Cr(Ⅳ)的离子形态及-NH2有关,通过吸附剂与吸附质之间的电子共用或静电吸附来实现的。而Fe3O4@SiO2@mSiO2的吸附过程则只是通过介孔结构实现简单的物理吸附。对于吸附重金属离子而言,除了需要发达的介孔结构外,合适的官能团修饰也是提高重金属离子吸附性能的关键。
[1]LI Guang-Lu(李广录),HE Tao(何涛),LI Xue-Mei(李雪梅). J.Prog.Chem.(化学进展),2011,23(6):1081-1089
[2]Laurent S,Forge D,Port M,et al.Chem.Rev.,2008,108: 2064-2110
[3]WANG Miao-Miao(王苗苗),LI Qun-Yan(李群艳),WEI Qi (韦奇),et al.Chem.J.Chinese Universities(高学校化学学报),2013,34(2):299-305
[4]XIONG Shan(熊珊),JIANG Xiang-Ping(江向平),LI Ju-Mei (李菊梅),et al.J.Chin.Ceram.Soc.(硅酸盐学报),2015,43 (7):946-951
[5]LIU Yun-Fang(刘云芳),REN Sen(任森),WU Ri-Liang (吴日良),et al.Chinese J.Inorg.Chem.(无机化学学报), 2015,31(12):2373-2378
[6]Park M,Seo S,Lee I S,et al.Chem.Commun.,2010,46: 4478-4480
[7]Wu P G,Zhu J H,Xu Z H.Adv.Funct.Mater.,2004,14(4): 345-351
[8]Liu Y,Fu R,Sun Y,etal.Appl.Surf.Sci.,2016,369:267-276
[9]Morel A L,Nikitenko S I,Gionnet K,et al.ACS Nano,2008, 2(5):847-856
[10]Stjerndahl M,Andersson M,Hall H E,et al.Langmuir, 2008,24(7):3532-3536
[11]Zhao W R,Gu J L,Zhang L X,et al.J.Am.Chem.Soc., 2005,127(25):8916-8917
[12]Fu R,Jin X M,Liang J L,et al.J.Mater.Chem.,2011,21: 15352-15356
[13]WANG Rong(汪绒),HAN Hai-Zhou(韩海洲),ZHENG Xing-Wang(郑行望),et al.Acta Chim.Sinica(化学学报), 2010,68(17):1726-1734
[14]SHEN Bin(沈彬),WANG Yong-An(王永安),WANG Zhi-Fei (王志飞),et al.Acta Phys.-Chim.Sin.(物理化学学报), 2010,26(7):1860-1866
[15]Bao S,Tang L,Li K,et al.J.Colloid Interface Sci., 2016,462:235-242
[16]Yang J,Shen D,WeiY,etal.Nano Res.,2015,8(8):2503-2514
[17]Tan H,Xue J M,Shuter B,et al.Adv.Funct.Mater.,2010, 20:722-731
[18]Chinese Environmental Protection Bureau(国家环保局). Analytical Method for Monitoring Water and Waste Water (水和废水监测分析方法).Beijing:China Environmental Science Press,2002:344-346
[19]Li Y,Sun J,Zhang L,et al.Appl.Surf.Sci.,2012,258: 3333-3339
[20]Panella B,Vargas A,Baiker A.J.Catal.,2009,261:88-93
[21]Baby T T,Ramaprabhu S.Talanta,2010,80:2016-2022
[22]Gai S,Yang P,Li C,et al.Adv.Funct.Mater.,2010,20:1-7
[23]XUE Hong-Tao(薛红涛),SHEN Shui-Fa(沈水发),PAN Hai -Bo(潘海波),et al.J.Inorg.Mater.(无机材料学报), 2009,24(3):577-580
[24]Nagao D,Satoh T,Konno M.J.Colloid Interface Sci., 2000,232(1):102-110
[25]Weng C H,Wang J H,Huang C P.Water Sci.Technol., 1997,35:55-62
[26]Ho Y S,McKay G.Proc.Biochem.,1999,34(5):451-465
Preparation and Adsorption Properties of Amino Modified Magnetic Mesoporous Microsphere Fe3O4@SiO2@mSiO2
XUE Juan-Qin*,1XU Shang-Yuan1ZHU Qian-Wen1QIANG Liang-Sheng2MA Jing*,1
(1Department of Chemical Engineering,Xian University of Architecture and Technology,Xi′an 710055,China)
(2School of Chemistry and Chemical Engineering,Harbin Institute of Technology,Harbin 150001,China)
The SiO2encapsulated Fe3O4composite magnetic microsphere was synthesized by a modified Stöber method with Fe3O4nanoparticles as cores.The structures and properties of the composite were characterized by XRD,SEM,TEM,N2absorption-desorption,FTIR and VSM.The results showed that the as-prepared magnetic microspheres exhibited a spherical shape and uniform particle size distribution.This material possessed a BET surface area of 413 m2·g-1and saturation magnetization of 68.93 emu·g-1,respectively.The particle morphology became more uniform with the increase of TEOS volumes when silica shell became thicker.The saturation magnetization of the nanocomposite particles decrease with the thickness of the silica shell increasing,the coercive force of particles was almost unchanged,and remained favorable to superparamagnetism.Based on the composite magnetic microsphere,the new magnetic nano-adsorbent Fe3O4@SiO2@mSiO2-NH2was fabricated by coating silica from a covalent binding of amino(-NH2)onto the surface of Fe3O4@SiO2@mSiO2microsphere.And the adsorption properties of the adsorbent for Cr(Ⅳ)ions were investigated.The study on kinetics of Cr(Ⅳ)showedthat the adsorption reaction could be considered as the pseudo-second-order kinetic model.The adsorption capability was significantly affected by the distributions of Cr(Ⅳ)species and-NH2.The adsorption mechanisms of Cr(Ⅳ)including both ion exchange and electrostatic gravitation were also discussed.
core-shell structure;composite magnetic microsphere;mesoporous silica;absorption property
X132.2;TB333
A
1001-4861(2016)09-1503-09
10.11862/CJIC.2016.215
2016-05-10。收修改稿日期:2016-08-07。
国家自然科学基金青年基金(No.51402231)、教育部高等学校博士学科点专项科研基金(No.20136120120017)、陕西省科技厅自然科学项目(No.2014JQ2076)和陕西省2015年大学生创新创业训练计划项目(No.1153)资助。
*通信联系人。E-mail:huagong1985@163.com,majing@xauat.edu.cn;会员登记号:S06N8536M1605。
猜你喜欢
发光学报(2021年7期)2021-07-23
潍坊学院学报(2020年6期)2020-11-22
汽车文摘(2018年1期)2018-11-26
环境科技(2017年6期)2018-01-17
组织工程与重建外科杂志(2018年6期)2018-01-12
中成药(2017年6期)2017-06-13
西安工程大学学报(2016年6期)2017-01-15
发光学报(2016年10期)2016-11-19
天然产物研究与开发(2016年6期)2016-06-05
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
