
固相萃取净化-超高效液相色谱串联质谱测定水产品中氯硝柳胺
2016-12-14 12:16严忠雍李佩佩
广州化工 2016年22期
严忠雍,祝 银,方 益,陈 思,李佩佩,龙 举
(浙江省海洋水产研究所,浙江省海洋渔业资源可持续利用技术研究重点实验室,浙江 舟山 316021)
固相萃取净化-超高效液相色谱串联质谱测定水产品中氯硝柳胺
严忠雍,祝 银,方 益,陈 思,李佩佩,龙 举
(浙江省海洋水产研究所,浙江省海洋渔业资源可持续利用技术研究重点实验室,浙江 舟山 316021)
建立了水产品中氯硝柳胺的固相萃取净化-液相色谱串联质谱分析方法。样品经2%氨水乙腈溶液提取,弗罗里硅土柱净化,液相色谱串联质谱分析。在ACQUITY UPLC BEH C18色谱柱上进行分离,采用梯度洗脱,电喷雾正离子电离,多反应监测模式,外标法定量。结果表明,氯硝柳胺在2.5~100 μg/L浓度范围内线性相关系数均大于0.999,对应的检出限和定量限均为0.2 μg/kg和0.5 μg/kg,平均回收率为86.8%~91.3%;相对标准偏差为3.14%~6.74%。本方法基质干扰小、净化效果强、灵敏度高,适合水产品中氯硝柳胺的分析测定。
氯硝柳胺;固相萃取;液相色谱串联质谱
氯硝柳胺(Niclosamide,NIC)是一种新型人工合成的高效灭绦虫药,为水杨酰胺类衍生物,其抗虫机制为抑制虫体细胞内线粒体氧化磷酸化过程,能量物质ATP生成减少,使绦虫的头节和邻近节片变质,虫体从肠壁脱落随粪便排出体外[1-4]。可用作抗蠕虫药和杀软体动物剂,驱除猪牛等动物体内的绦虫,也可以灭螺,杀鳗。在水产行业中,主要用于防治血吸虫,同时对鱼类和两栖类动物毒性较大,因而在鱼溏换新水之前被作为清塘剂来杀灭野杂鱼。由于其用药方式为直接对水体泼洒用药,引起的水生动物急性死亡事件有所增加,因此该药物在鱼肉中存在富集的潜在风险,并且是否通过食物链的传递最终积累到消费者而产生“三致”作用逐步引起人们的重视。
目前,国内外对氯硝柳胺的检测方法主要有电位滴定法、液相色谱法[5-6]、气相色谱串联质谱法[7]、液相色谱串联质谱法[8-10]等。电位滴定法专一性差,且易受人为操作影响;液相色谱法受样品基质影响大,且易出现假阳性;气相色谱串联质谱法虽也能有效测定氯硝柳胺,但应用程度低,不适合实际检测推广;液相色谱串联质谱法灵敏度高,精确度好,是目前最适合检测氯硝柳胺残留的分析方法。本方法利用固相萃取净化技术对样品进行前处理净化,并采用液相色谱-串联质谱法对氯硝柳胺进行同时检测,建立一种简单、高效、灵敏测定水产品中性激素残留量的分析方法。
1 实 验
1.1 材料与试剂
乙酸铵、甲酸(色谱纯),美国sigma公司;乙腈(色谱纯),德国Merck公司;氨水(分析纯),上海国药试剂有限公司;实验用水为Millipore-Q系统制备的超纯水;氯硝柳胺标准品(纯度均≥98%),德国Dr.Ehrenstorfer公司;其他试剂均为分析纯。
标准储备液:准确称取5.00 mg氯硝柳胺标准品,用乙腈溶解定容于50 mL容量瓶,-18 ℃避光保存,保存期限为2个月。使用时用乙腈逐级稀释至所需浓度。
1.2 仪器与设备
ACQUITYTMUPLC-Quattro Premier XE质谱仪(配电喷雾离子源),美国Waters公司; MS3 Digital漩涡混合器,德国IKA公司;T18匀浆机,德国IKA公司;N-EVAP112氮气吹干仪,美国Organomatio公司;R-125旋转蒸发浓缩仪,瑞士Buchi公司;超声波清洗器,上海科导超声仪器有限公司;SPE-12固相萃取装置,美国supelco公司;Centrifuge5810高速离心机,德国Eppendorf公司;弗罗里硅土柱(1 g,6 mL),上海安谱科学仪器有限公司。
1.3 方法
1.3.1 样品提取
准确称取样品(5.00±0.02) g于50 mL具塞离心管中,加2%氨水乙腈溶液15 mL,涡旋振荡1 min,20 ℃超声提取15 min,7 000 r/min离心5 min,上清液移至梨形瓶中,残渣再用15 mL氨化乙腈重复提取一次,合并上清液于梨形瓶中,30℃旋转蒸发至1.0 mL,加2mL乙腈溶解备用。
1.3.2 样品净化
将提取浓缩液全部过已经预先用3 mL乙腈活化的弗罗里硅土柱,3 mL二氯甲烷洗脱萃取柱,并收集洗脱液,于30 ℃氮气吹干,最后用1 mL初始流动相定容,经0.22 μm滤膜后,按工作条件进行仪器分析。
1.4 仪器工作条件
色谱柱:ACQUITY UPLC BEH C18柱(2.1 mm×50 mm,1.7 μm);样品室温度6 ℃;柱温35 ℃;进样量10 μL;流动相A为含0.1%甲酸的2 mM乙酸铵溶液,B为乙腈,流速0.2 mL/min;梯度洗脱见表1。
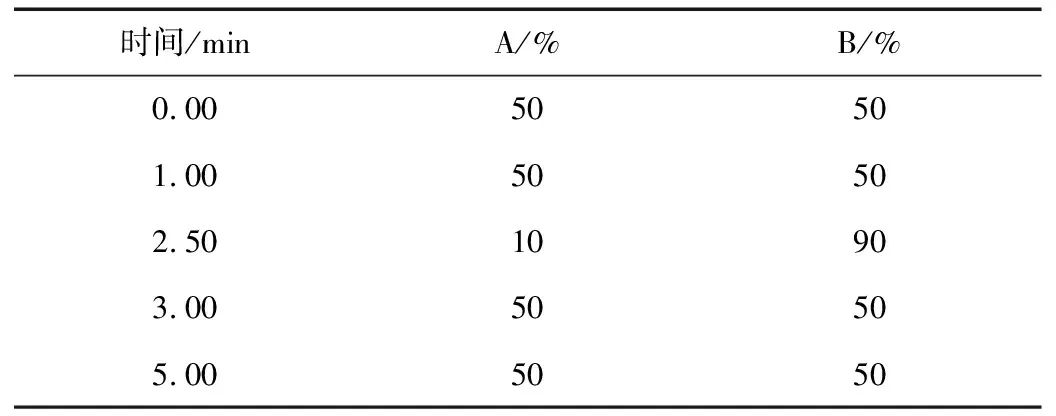
表1 梯度洗脱程序
质谱条件:电喷雾离子源,负离子扫描(ESI-);检测方式:多反应监测模式(MRM);毛细管电压:3.0 kV;离子源温度:115 ℃;脱溶剂气温度:370 ℃;锥孔气流量:60 L/h;脱溶剂气流量:700 L/h;分析物母离子及子离子、锥孔电压、碰撞能量等质谱条件参数如表2所示。

表2 氯硝柳胺的质谱条件参数
*定量离子。
2 结果与讨论
2.1 提取剂的选择
为达到良好的提取效果,实验比较了乙腈、氨化乙腈、酸化乙腈、甲醇和乙酸乙酯作为提取剂时对氯硝柳胺的提取效率。通过采用不同的提取剂对阴性加标样进行萃取,考察比较对应的氯硝柳胺的回收率,筛选出最适合的提取剂。实验结果表明,2%氨水乙腈对应的回收率最高,对氯硝柳胺的提取效率最好,选择2%氨水乙腈作为本实验提取剂。
2.2 色谱质谱条件优化
实验比较了氯硝柳胺在对应有机流动相为乙腈与甲醇时的响应效果,发现氯硝柳胺在乙腈中有更大的响应值,更有利于色谱分析判定,降低检出限,因此选择乙腈作为有机相。乙酸铵通常作为液相色谱分离水相中的常用缓冲盐,用以提高水相的稳定性,且能提高质谱的离子化效率,减少由于流动相的不稳定性导致色谱峰漂移的可能性,确保实验的批间准确性。甲酸能通过改变流动相的pH提高色谱分离效果,改善分析物的峰形,故最终选择含0.1%甲酸的2 mM乙酸铵溶液作为水相。氯硝柳胺在实验开发的梯度洗脱下,将单个样品分析时间减少至5 min,提高了仪器的分析效率,如图1所示。
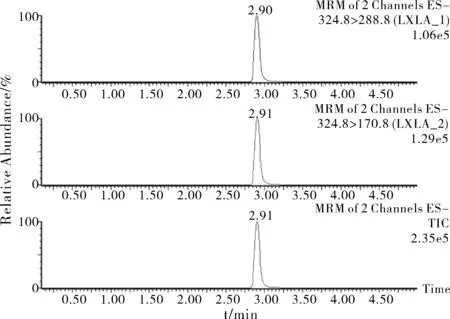
图1 氯硝柳胺的MRM图
为获得氯硝柳胺的质谱条件参数,实验分别在ESI+和ESI-扫描模式下查找比较氯硝柳胺的响应。结果表明,ESI-扫描模式下氯硝柳胺的响应值较高,因此在ESI-模式下查找氯硝柳胺的特征离子。在一级全扫描下,发现特征离子峰[M-H]-(m/z=324.8)丰度最高,选择其作为氯硝柳胺的母离子;通过母离子裂解的离子碎片信息,发现氯硝柳胺对应的离子碎片峰m/z=170.8和m/z=288.8丰度较其他碎片峰高,故选择依次作为定量离子和定性离子。
2.3 基质效应、线性关系和检出限
为考察实验的基质效应,将阴性样品提取液作为基质溶液,分别用初始流动相和基质溶液各配制5和20 μg/L氯硝柳胺溶液,比较对应的平均峰面积。通过计算得到基质效应(ME)为4.9%,表明本实验条件下基质效应影响较小,满足分析测试的需要。
用氯硝柳胺标准液配制浓度为2.5、5.0、25、50.0和100.0 μg/L标准工作溶液,以氯硝柳胺浓度为横坐标,其对应的峰面积为纵坐标,制作标准曲线,如图2所示。结果表明,氯硝柳胺在浓度为2.5~100.0 μg/L范围内线性关系良好。分析方法的理论检出限为被测物质丰度较弱子离子的信号/噪声比(RSN)不小于3,定量限为信噪比不小于10,将加标浓度逐级稀释测定信噪比,最终确定本方法的检出限为0.2 μg/kg,定量限为0.5 μg/kg。
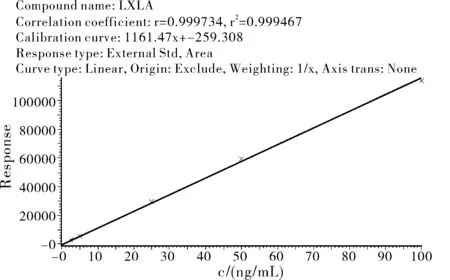
图2 氯硝柳胺标准曲线
2.4 回收率及精密度

表3 氯硝柳胺在阴性鲫鱼中加标回收率和相对标准偏差(n=6)
称取5.00 g阴性鲫鱼样品,添加1.00,5.00和20.0 μg/kg 3种氯硝柳胺加标浓度,每个加标水平重复测定6次,计算对应的准确度和精密度。结果如表3所示。由表3可知,氯硝柳胺的平均回收率为86.8%~91.3%;相对标准偏差为3.14%~6.74%;满足氯硝柳胺残留检测分析的要求。
2.5 样品分析
为了验证本方法的适用性和实用性,采用本方法对市场采集的15份水产样品进行检测分析,被测样品中氯硝柳胺的含量均低于0.5 μg/kg。表明此批次抽检样品均未有氯硝柳胺检出,氯硝柳胺在水产品行业中得到了有效的控制,但仍需严控水产品的质量安全。
3 结 论
实验建立了固相萃取柱净化水产品中氯硝柳胺的超高效液相色谱串联质谱分析方法,利用弗罗里硅土柱吸附净化样品,降低样品基质的干扰,结合超高效液相-串联质谱仪的强分离性和高灵敏度,提高了样品回收率,极大降低检出限和定量限,有利于日常检测工作的推广使用。
[1] 丁力,吕昌银,冯家力,等. 洞庭湖区田鼠肝脏组织中氯硝柳胺的液相色谱-质谱/质谱检测方法研究[J]. 中国卫生检验杂志,2007(12):2153-2155.
[2] 姜友富,王俊,嵇正平,等. 氯硝柳胺现场检测方法和检测仪的研究与开发[J]. 中国血吸虫病防治杂志,2009(03):209-211.
[3] 程梦馨,赵俊章,王晓兵,等. 氯硝柳胺对人结肠癌SW480细胞生长及凋亡的影响[J]. 武汉大学学报:医学版,2015(06):906-908, 912.
[4] Dai J R, Wang W, Liang Y S, et al. A novel molluscicidal formulation of niclosamideg[J]. Parasitology Research, 2008, 103(2): 405-412.
[5] 严相平,王康蕊,王浦海,等. HPLC法测定泥土中氯硝柳胺的含量[J]. 农药,2011(05):350-351.
[6] 王娇娜,龙海,肖同,等. HPLC测定氯硝柳胺的含量[J]. 华西药学杂志,2006(04):401-402.
[7] 石琳,黄金良,刘然,等. 氯硝柳胺的GC/MS检验[J]. 刑事技术,2013(06):51-53.
[8] 万译文,黄向荣,李小玲,等. 高效液相色谱-质谱/质谱联用法检测渔业水体中氯硝柳胺[J]. 分析试验室,2015(01):53-56.
[9] 伍一红,龚道新,彭筱,等. 高效液相色谱法分析水稻和稻田中的氯硝柳胺乙醇胺盐残留[J].色谱,2011(11):1098-1102.
[10]周颖,周艺彪,何明祯,等. 超声萃取-高效液相色谱测定土壤中的氯硝柳胺[J].中国血吸虫病防治杂志,2009(01):35-38.
Determination of Niclosamide in Aquatic Products Based on Solid Phase Extraction Coupled with Liquid Chromatography Tandem Mass Spectrometry
YANZhong-yong,ZHUYin,FANGYi,CHENSi,LIPei-pei,LONGJu
(Marine Fisheries Research Institute of Zhejiang, Key Lab of Sustainable Utilization of Technology Research for Fishery Resource of Zhejiang, Zhejiang Zhoushan 316021, China)
A new method of solid phase extraction combined with high liquid chromatography-tandem mass spectrometry was established for the determination of niclosamide in aquatic products. The sample was extracted with 2% ammonia acetonitrile solution, cleaned up by florisil coupled with high performance liquid chromatography-mass spectrometry. The chromatographic separation was performed on an ACQUITY UPLC BEH C18column with gradient elution. External standard method was used for quantification and the positive electrospray ionization source under the multiple reaction monitoring mode. Linear ranges of niclosamide was in the range of 2.5~100 μg/L with correlation coefficient more than 0.999. The limit of detection (LOD) and limit of quantification (LOQ) for niclosamide was calculated as 0.2 and 0.5 μg/kg. The recoveries of analyte in sample were in the range of 86.8%~91.3% with the relative standard deviation of less than 6.74%. Therefore, the method could be used to identify and quantify niclosamide in aquatic products with less matrix interference and satisfactory sensitive.
niclosamide; solid phase extraction; high performance liquid chromatography-mass spectrometry
浙江省科研院所公共科技服务项目(2016F30022)。
O656.31
B
1001-9677(2016)022-0081-03
严忠雍(1990-),男,助理工程师,学士,主要从事渔业环境监测和水产品质量安全研究。
猜你喜欢
煤化工(2022年3期)2022-07-08
今日农业(2021年4期)2021-11-27
今日农业(2021年15期)2021-11-26
色谱(2021年7期)2021-06-07
渔业致富指南(2019年21期)2019-11-21
新世纪智能(英语备考)(2018年11期)2018-12-29
中国环境监察(2016年7期)2016-10-23
中国现当代社会文化访谈录(2016年0期)2016-09-26
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
