
载能碳离子撞击石墨烯中Stone-W ales缺陷的动力学研究
2016-12-01 01:31张超王东琪孟祥瑞潘成岭吕思远安徽理工大学材料科学与工程学院淮南00安徽理工大学能源与安全学院淮南00中国科学院高能物理研究所多学科中心北京00049
无机化学学报 2016年1期
张超 王东琪 孟祥瑞 潘成岭 吕思远(安徽理工大学材料科学与工程学院,淮南00)(安徽理工大学能源与安全学院,淮南00)(中国科学院高能物理研究所多学科中心,北京00049)
载能碳离子撞击石墨烯中Stone-W ales缺陷的动力学研究
张超1,2王东琪*,3孟祥瑞2潘成岭*,1吕思远1
(1安徽理工大学材料科学与工程学院,淮南232001)
(2安徽理工大学能源与安全学院,淮南232001)
(3中国科学院高能物理研究所多学科中心,北京100049)
采用分子动力学模拟方法,研究了载能碳离子撞击石墨烯中Stone-Wales缺陷的动力学过程,计算了Stone-Wales缺陷中初级碰撞原子的离位阈能和载能碳离子使其移位的入射阈能,并与完美石墨烯结构计算结果进行对比。通过分析初级碰撞原子与入射离子动能和势能随时间的变化关系,研究了碰撞过程中能量转移过程。研究结果表明,初级碰撞原子产生离位并最终脱离石墨烯体系的最小能量为25.0 eV。当初始动能为23.0 eV时,Stone-Wales缺陷中2个七元环共用的碳-碳键旋转90°形成了完美的石墨烯结构。此外,还发现使Stone-Wales缺陷中初级碰撞原子发生离位的载能碳离子最小入射能为41.0 eV。
石墨烯;分子动力学;Stone-Wales缺陷;碰撞;移位阈能
0 引言
石墨烯是由碳原子按照六边形进行排布的二维晶体[1],由于独特的各向异性结构和杰出的力学性能、奇特的导电性能、优异的光学性能以及相对论量子效应[2-4],使其在许多领域中有着广泛的应用,诸如晶体管[5-6]、太阳能电池[7]、传感器[8-9],以及可用于制备复合材料,储氢材料和高能电容器等材料[10]。在这些应用环境中,石墨烯基电子器件由于会遭受荷能粒子的辐照而导致性能发生变化。对此,许多研究者已从实验和理论上分别研究了载能粒子对石墨烯的材料改性工作,以探索石墨烯在荷能粒子辐照下结构和性能变化[11-15]。石墨烯中已发现的缺陷构型有空位型缺陷[16]、杂质原子缺陷、Stone-Wales(SW)缺陷[17]以及5-7对缺陷构型[18]等。由于SW缺陷广泛存在于碳纳米材料以及其它具有sp2杂化的共价键结构材料中,例如SiC纳米管[19],而且,辐照引起的SW缺陷能够增强石墨层卷曲成碳纳米管和富勒烯的趋势,并且SW缺陷对石墨烯结构的本征皱曲有着重要作用[10],所以,研究SW缺陷对深入探究石墨烯结构变化和性能改进,以及对石墨烯基电子器件的广泛使用具有重要理论意义和工程应用价值。此外,Lehinen等[20]利用分析势和密度泛函理论相结合方法模拟了载能离子对石墨烯的辐照损伤行为,重点研究了石墨烯中缺陷的类型和浓度变化。然而,对于石墨烯晶体中结构变化的动力学过程并未见相关报道。我们曾利用分子动力学方法,研究了载能粒子碰撞石墨烯的动力学过程,研究了石墨烯中靶原子的移位阈能,观察到单空位、双空位和SW缺陷构型的演化过程[21]。然而,该研究仅对一次辐照下石墨烯的辐照损伤行为进行了研究,由于在辐照损伤过程中也会发生二次辐照,即一次辐照形成的缺陷构型受到载能粒子的继续辐照而形成了新的缺陷构型。本文将在上述研究基础上,研究载能碳离子对石墨烯中SW本征缺陷的辐照损伤行为,重点研究SW缺陷中初级碰撞原子的离位阈能和载能碳离子使其移位的入射阈能,探究石墨烯中SW本征缺陷的抗辐照能力,希望对石墨烯改性,以及对石墨烯基功能材料提供理论依据。
1计算模型与方法
1.1初始结构
本文首先建立具有Stone-Wales本征缺陷的石墨烯模型,X和Y方向的长度分别为2.2和2.6 nm;采用最小梯度法对结构进行优化,在优化后的结构中选取出SW缺陷中典型原子(如图1箭头所指原子)作为初级碰撞原子(PKA)。为了获得最小的辐照能量,本文根据毛飞等[21]研究石墨烯的辐照损伤报道,发现对头碰撞时所需入射能量最小。因此,在模拟载能粒子与靶材料的碰撞过程中,本文仅仅考虑入射粒子与PKA的对头碰撞,故将入射碳原子建立在PKA上方且垂直于石墨烯平面方向上,入射粒子与PKA的初始距离保持在2.0 nm以上,以避免初始状态具有相互作用。
1.2原子间势函数
在模拟计算中,体系所有原子间相互作用均采用AIREBO势函数[22];其次,为了描述高能粒子间短程相互作用,采用ZBL势函数[23]。由于入射粒子能量较低,在碰撞过程中核阻止将起主要作用,所以本文计算中不考虑电子阻止对辐照损伤的影响。此外,为了与实验进行类比,同时,由于低电荷量的电荷效应可以忽略,所以本工作中不考虑电荷效应对碰撞过程的影响,即本文中入射碳原子称为入射碳离子。
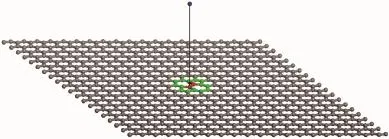
图1石墨烯中SW缺陷模拟示意图(图中蓝色球表示入射碳离子,箭头所指球为初级碰撞原子(PKA),绿色球所围区域表示SW缺陷区域)Fig.1 Schematic representation of the simulation setup of SW defect in graphene(Blue sphere indicates the incident carbon ion; The red sphere,which is pointed with an arrow,indicates the primary knock-on atom(PKA);The area surrounded by green spheres represents SW defect)
1.3模拟细节介绍
在辐照模拟过程中,本文采用NVE(等原子数、等体积、等能量)系综,时间步长取0.01 fs,模拟总步数取2.0×105步。模拟测试发现系统总能量的均方根误差(RMSE)约为4.9×10-2eV,几乎是入射动能的0.1%,这表明碰撞过程中系统的总能量守恒。辐照模拟结束后,采用NVT系综对体系进行弛豫50 ps,时间步长为1 fs,系统温度控制在300 K。为了得到稳定的缺陷构型,将对弛豫后的结构进行退火模拟,使系统温度由300 K先升温至1 500 K,然后再退火至300 K,升温和退火的模拟时间均为50 ps。为防止计算模拟过程中石墨烯体系发生移动,在计算过程中采用固定质心方法,即每运行一个时间步长,通过平移整个体系,使质心调整到初始位置。
本文计算模拟过程是由开源的分子动力学计算程序LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)[24]完成。为进一步分析结构变化,使用了可视化软件VMD[25]。此外,为了反映原子间成键情况,采用密度泛函基础的紧束缚(DFTB)方法[27]计算了模拟后石墨烯结构的电荷密度分布情况。
为了验证计算模型的准确性,本文首先利用该模型计算了石墨烯中单空位缺陷的形成能为7.5 eV,该值与采用第一性原理的计算结果7.7 eV能够较好地吻合[26];此外,计算了完美无缺陷石墨烯中靶原子离位阈能为25.0 eV,该值与Krasheninnikov等[27]采用密度泛函基础的紧束缚(DFTB)方法计算结果(22.5 eV)基本一致,这些结果表明,本工作所采用的计算模型能够较为准确地描述石墨烯体系的碰撞动力学过程。
2 结果与讨论
2.1Stone-W ales缺陷中初级碰撞原子离位阈能和能量转移过程
首先,赋予PKA初始动能(Ek),使该动能的速度方向垂直于石墨烯平面,当赋予PKA初始动能低于23.0 eV时,实验观察到初级碰撞原子先是离开晶格中原来位置,随后又恢复至原来位置,最终体系仍然保持SW缺陷构型,结构不发生任何变化。当Ek= 23.0 eV时,PKA仍然不能脱离石墨烯体系,但能够使SW缺陷构型转变为无缺陷石墨烯结构,具体动力学演化过程如图2所示。当模拟时间t=15 fs时,原子1与原子3和5之间的化学键发生断裂,同时原子2与原子4之间化学键也发生断裂,如图2(b)所示。随着演化的进行,当t=150 fs时,原子2和5、原子1和3分别形成了新的化学键,并且原子2与4之间也重新形成了化学键,如图2(c)所示。当t= 2 000 fs时,从图中可以观察到,原子1和4之间形成了新的化学键,同时原子2和4之间化学键发生断裂。从最后的构型中可以看出,最终结构为完美无缺陷的石墨烯结构,如图2(d)和2(e)所示。整个演化动力学过程相当于将原子1和2之间的化学键逆时针旋转90°得到。为了得到稳定结构,本文对体系进行升温退火模拟,结果发现,升温退火后(t=152 ps),体系结构仍然保持不变,为完美的石墨烯构型。
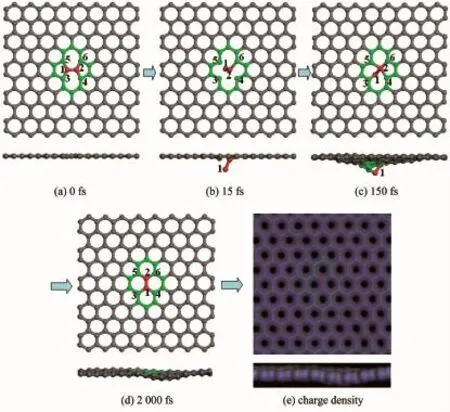
图2 PKA初始动能为23.0 eV时原子演化过程和最终电荷密度的俯视图和侧视图(红色球1表示PKA)Fig.2 Atomic processes and the final charge density from the top view and side view with the incident energy of 23.0 eV for PKA(The red sphere numbered as 1 indicates PKA)
当PKA初始动能达到25.0 eV时,PKA将有足够能量克服离位势垒,最终脱离石墨烯体系。具体演化过程如图3所示。从图3(b)可以看出,当模拟时间t=10 fs时,原子1与原子2、3和5之间的化学键全部发生断裂。当模拟时间t=2 000 fs时,原子1已经脱离石墨烯体系的束缚,石墨烯体系中原子2、3和5均存在不饱和的悬挂键,如图3(c)所示。通过对体系进行升温退火模拟,结果发现,最终原子2和5之间形成了新的化学键,变为5-9缺陷构型,如结构图3(d)和对应的电荷密度图3(e)所示。
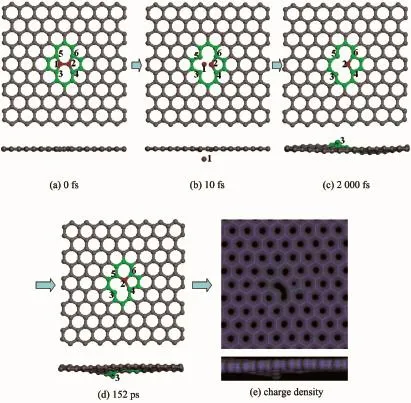
图3 PKA初始动能为25.0 eV时原子演化过程和最终电荷密度的俯视图和侧视图(红色球1表示PKA)Fig.3 Atomic processes and the final charge density from the top view and side view with the incident energy of 25.0 eV for PKA(The red sphere numbered as 1 indicates PKA)
为了研究模拟过程中PKA能量转移过程,本文分别计算了Ek=23.0 eV和25.0 eV时,PKA的动能和势能随时间的演化关系,如图4所示。从图4(a)可以看出,PKA初始动能为23.0 eV,初始势能约为-6.6 eV。随着演化进行,PKA动能先缓慢降低,然后在t=5 fs时快速下降;当t=16 fs时,Ek已降至约3.0 eV;随后,PKA动能略微反弹后又继续降低,当t= 300 fs时,PKA动能降至约0.1 eV,这表明PKA动能几乎完全耗散至石墨烯体系。从PKA势能曲线可以观察到,其势能先是缓慢增加,当t=5 fs以后,Ep快速增加,在t=9 fs时,已增加到最大值。这表明该阶段PKA正在试图离开晶格中原来位置,导致势能增加,该结果可以从图2(b)原子动力学演化过程中清楚地观察到,原子1和原子3、5之间化学键发生断裂,并带动原子2向垂直于石墨烯平面方向移动。继续观察图4(a)中势能曲线发现,在t=10 fs附近,Ep略微下降,并在130 fs附近再次下降,最终当t= 2 000 fs时,Ep降为约-7.4 eV,此时对应于图2(d)中的结构图,PKA与原子3和4形成了新的化学键。
当PKA初始动能为25.0 eV时,其动能和势能演化曲线如图4(b)所示。从图中可以看出,PKA动能曲线起初缓慢下降,势能曲线缓慢上升,当模拟时间t=4 fs以后,PKA动能快速下降,当t=13 fs时已降至约7.8 eV。同时,PKA势能逐渐上升,在t=10 fs时上升至-0.1 eV,最终PKA势能变为0.0 eV,表明PKA已经脱离石墨烯体系,如原子结构演化图3(c)所示。此外,从动能曲线可以看出PKA脱离石墨烯体系残留的动能约为7.7 eV。

图4 PKA动能和势能随时间演化关系,图(a)和(b)分别表示初始能量为23.0 eV和25.0 eVFig.4 Time evolutions of the kinetic energies and potential energies of the PKA at incident energies of 23.0 eV (a)and 25.0 eV(b),respectively
2.2载能碳离子对Stone-W ales缺陷辐照阈能与能量转移过程
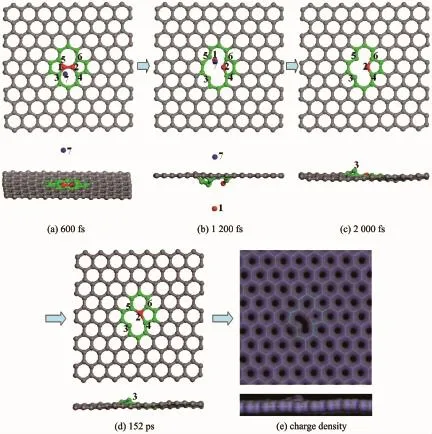
图5入射离子能量为41.0 eV时碰撞动力学过程和最终电荷密度的俯视图和侧视图(蓝色和红色(1)球分别表示入射碳离子和初级碰撞原子)Fig.5 Time evolution of the collision and the final charge density from the top view and side view with the incident energy of 41.0 eV for the incident ion(Blue and red(numbered as 1)spheres indicate the incident carbon ion and the PKA,respectively)
为了更好地了解SW缺陷的抗辐照性能,本工作研究了载能碳离子辐照SW缺陷的动力学过程。首先赋予入射碳离子一初始动能(Ein),动能速度方向垂直于石墨烯平面。模拟结果发现,当Ein<40.0 eV时,PKA先是离开原来位置,但随后又立即恢复至原来位置,SW结构不发生任何变化。当Ein=41.0 eV时,原子演化动力学过程如图5所示。从图中可以发现,当模拟时间t=1 200 fs时,PKA与原子2、3和5之间化学键发生断裂而离开了原来位置,如图5(b)所示。当t=2 000 fs时,PKA已经脱离石墨烯体系,如图5(c)所示。通过对碰撞后体系进行退火模拟发现,最终体系演化为一个五元环和一个九元环(5-9)的缺陷构型,如图5(d)和对应的电荷密度图5(e)所示。此外,在碰撞过程中还发现入射碳离子与石墨烯体系相互作用后受到斥力作用而反弹。
图6展示了入射碳离子与初级碰撞原子动能和势能随时间的演化关系。从图6(a)可以看出,入射离子动能在模拟时间t=76 fs以后快速下降,势能略微上升。当t=80 fs时,入射离子动能已降至约0.3 eV,随后又缓慢增加至1.9 eV并保持不变,这表明入射离子已经远离石墨烯体系。该结论也可以从势能值最终变为0.0 eV而得到。同时,从图5(c)结构图可以发现,此时入射离子也已经离开石墨烯体系。对于PKA能量变化情况,如图6(b)所示,在t=76 fs以后,PKA势能快速增加,并于t=78 fs时,势能上升至最大值5.7 eV,这是由于PKA受到入射离子碰撞而产生强大的斥力所致。随着演化进行,PKA势能缓慢下降,最终降为0.0 eV,表明此时PKA已经脱离石墨烯体系束缚,这与图5(c)结构图一致,图中显示PKA和入射离子均已远离石墨烯体系。从图6(b)中PKA动能变化曲线可以发现,在t=77 fs以后,Ek快速上升,并于81 fs时,Ek上升至最大值约33.3 eV,随后又快速下降,最终降至约7.9 eV,即为PKA脱离石墨烯体系残余的动能。
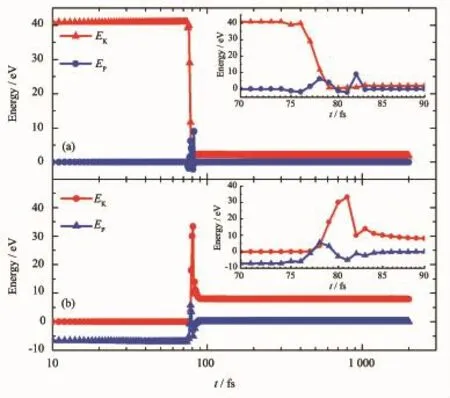
图6入射离子(a)与PKA(b)动能和势能随时间的演化关系(其中插图是对模拟时间为70~90 fs范围内曲线放大效果图)Fig.6 Time evolutions of the kinetic energies and potential energies of the incident ion(a)and the PKA(b)at the incident energy of 41.0 eV,respectively(The insets in(a)and(b)show the close-up images of kinetic energy and potential energy in the time ranging from 70 to 90 fs)
3 结论
本文采用分子动力学方法,研究了石墨烯中Stone-Wales缺陷典型原子的离位阈能和使靶原子移位的入射碳离子入射阈能。结果表明,当初级碰撞原子能量低于23.0 eV时,Stone-Wales缺陷构型不发生任何改变,仍然保持原有结构;当PKA能量增至23.0 eV时,Stone-Wales缺陷中2个七元环共用的碳-碳键将旋转90°形成完美的六元环石墨烯结构;当PKA初始动能增至25.0 eV时,PKA将发生离位并最终脱离石墨烯体系,即Stone-Wales缺陷中典型原子的离位阈能为25.0 eV,这与完美石墨烯中靶原子的离位阈能一致。通过研究载能碳离子与Stone-Wales缺陷相互作用,结果发现,使Stone-Wales缺陷中典型原子发生离位的最小入射能约为41.0 eV。根据双体碰撞近似理论,入射能应为25.0 eV,两者差距较大的原因是因为载能碳离子与石墨烯体系相互作用过程中,入射离子不仅与PKA有相互作用,而且与PKA近邻的原子之间也有相互作用,从而导致载能离子的入射能既要传递部分能量给PKA,又要传递部分能量给PKA近邻的原子,使得最小入射能比采用双体碰撞模型计算的能量要高。本文研究结果将有助于利用离子辐照对石墨烯以及具有Stone-Wales本征缺陷的石墨烯结构进行调整,从而制备出抗辐射的以石墨烯为基体的纳米电子器件。
[1]Novoselov K S,Geim A K,Morozov S V,et al.Science,2004, 306:666-669
[2]Lee C,Wei X D,Kysar J W,et al.Science,2008,321:385-388
[3]LIU Xiao-Feng(刘晓峰),MI Chang-Huan(米常焕),ZHANG Wen-Qing(张文庆).Chinese J.Inorg.Chem.(无机化学学报), 2014,30(2):242-250
[4]JIN Lei(靳磊),FU Hong-Gang(付宏刚),XIE Ying(谢颖), et al.Chinese J.Inorg.Chem.(无机化学学报),2015,31 (3):446-451
[5]Liao L,Lin Y C,Bao M,et al.Nature,2010,467:305-308
[6]Lin Y M,Jenkins K A,Valdes-Garcia A,et al.Nano Lett., 2009,9:422-426
[7]Li X M,Zhu H W,Wang K L,et al.Adv.Mater.,2010,22: 2743-2748
[8]Cheng Z G,Li Q,Li Z J,et al.Nano Lett.,2010,10:1864-1868
[9]Rangel N L,Seminario J M.J.Chem.Phys.,2010,132: 125102
[10]Krasheninnikov A V,Nordlund K.J.Appl.Phys.,2010, 107:071301
[11]Ugeda M M,Brihuega I,Hiebel F,et al.Phys.Rev.B, 2012,85:121402
[12]Bellido E P,Seminario J M.J.Phys.Chem.C,2012,116: 4044-4049
[13]Mao F,Zhang C,Gao C Z,et al.J.Phys.:Condens.Matter., 2014,26:085402
[14]Teweldebrhan D,Balandin A A.Appl.Phys.Lett.,2009,94: 013101
[15]Åhlgren E H,Kotakoski J,Krasheninnikov A V.Phys.Rev. B,2011,83:115424
[16]Meyer J C,Kisielowski C,Erni R,et al.Nano Lett.,2008,8: 3582-3586
[17]Stone A J,Wales D J.Chem.Phys.Lett.,1986,128:501-503
[18]Gómez-Navarro C,Meyer J C,Sundaram R S,et al.Nano Lett.,2010,10:1144-1148
[19]Wang Z G,Gao F,Li J,et al.J.Appl.Phys.,2009,106: 084305
[20]Lehtinen O,Kotakoski J,Krasheninnikov A V,et al.Phys. Rev.B,2010,81:153401
[21]Mao F,Zhang C,Zhang Y,et al.Chin.Phys.Lett.,2012,29: 076101
[22]Stuart S J,Tutein A B,Harrison J A.J.Chem.Phys., 2000,112:6472
[23]Ziegler J F,Biersack J P,Littmark U.The Stopping and Range of Ions in Matter.New York:Pergamon,1985:21-25
[24]Plimpton S.J.Comput.Phys.,1995,117:1-19
[25]Humphrey W,Dalke A,Schulten K.J.Mol.Graphics,1996, 14:33-39
[26]Lehtinen P O,Foster A S,Ma Y,et al.Phys.Rev.Lett., 2004,93:187202
[27]Krasheninnikov A V,Banhart F,Li J X,et al.Phys.Rev.B, 2005,72:125428
Simulation Study of Collision Dynam ics of an Energetic Carbon Ion to the Stone-W ales Defect Site in Graphene
ZHANG Chao1,2WANG Dong-Qi*,3MENG Xiang-Rui2PAN Cheng-Ling*,1LÜ Si-Yuan1
(1School of Materials Science and Engineering,Anhui University of Science and Technology,Huainan,Anhui 232001,China) (2School of Energy and Safely,Anhui University of Science and Technology,Huainan,Anhui 232001,China)
(3Multidiscip linary Initiative Center,Institute of High Energy Physics,Chinese Academy of Sciences,Beijing 100049,China)
The collision dynamics processes of an energetic carbon ion to the Stone-Wales defect in graphene are investigated by using molecular dynamics method.We calculate the displacement threshold energy for the primary knock-on atom in Stone-Wales defect and the incident threshold energy for the projectile carbon ion prompting the target atom displacement,which are compared with the results of the perfect graphene.The energy transfer is studied by analyzing the time evolutions of the kinetic energies and potential energies of the primary knock-on atom and the incident ion.We find that the displacement threshold energy is 25.0 eV,which is the minimum kinetic energy for the primary knock-on atom to leave its original position and eventually escape from the graphene system.When the initial kinetic energy is 23.0 eV,the common C-C bond of the two heptagons in the Stone-Wales defect rotates 90°to form a perfect graphene structure.The minimum incident energy of the projectile required to drive the primary knock-on atom in the Stone-Wales defect to displace permanently from its original location is 41.0 eV.
graphene;molecular dynamics;Stone-Wales defect;collision;displacement threshold energy
O561.5
A
1001-4861(2016)01-0018-07
10.11862/CJIC.2016.025
2015-09-01。收修改稿日期:2015-10-27。
国家自然科学基金(No.11505003,21201006,21473206),安徽省自然科学基金,安徽省质量工程(No.2015jyxm132),安徽理工大学科研启动基金(No.ZX944),安徽理工大学国家自然科学预研基金(No.10029)和安徽省自然基金(No.1608085QA20)资助项目。
*通信联系人。E-mail:dwang@ihep.ac.cn,clpan@aust.edu.cn;Tel:010-88236606
猜你喜欢
——《势能》
文化纵横(2022年3期)2022-09-07
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
军民两用技术与产品(2022年1期)2022-06-01
金桥(2022年4期)2022-05-05
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
人大建设(2018年7期)2018-09-19
商周刊(2018年15期)2018-07-27
商周刊(2018年11期)2018-06-13
