
离子抑制色谱法测定注射用醋酸奥曲肽中乙酸的含量
2016-11-24 09:48:48李婧华刘桂霞李盼盼王智亮李卓荣山广志中国医学科学院医药生物技术研究所北京00050潍坊市人民医院山东潍坊604中国食品药品检定研究院北京00050
中国药房 2016年27期
李婧华,刘桂霞,李盼盼,王智亮,姚 静,李卓荣,山广志#(.中国医学科学院医药生物技术研究所,北京 00050;.潍坊市人民医院,山东潍坊 604;.中国食品药品检定研究院,北京 00050)
离子抑制色谱法测定注射用醋酸奥曲肽中乙酸的含量
李婧华1*,刘桂霞1,李盼盼1,王智亮2,姚静3,李卓荣1,山广志1#(1.中国医学科学院医药生物技术研究所,北京100050;2.潍坊市人民医院,山东潍坊261041;3.中国食品药品检定研究院,北京100050)
目的:采用离子抑制色谱法测定注射用醋酸奥曲肽中乙酸的含量,为药典标准的完善提供参考。方法:色谱柱为Rezex ROA-Organic Acid H+,流动相为0.002 5 mol/L硫酸溶液,流速为0.5 ml/min,检测波长为210 nm,柱温为45℃,进样量为100µl。结果:乙酸的检测质量浓度线性范围为0.394 4~78.89µg/ml(r=0.999 9);定量限为197.2 ng/ml,检测限为78.89 ng/ml;精密度、稳定性、重复性试验的RSD<2%;加样回收率为104.71%~109.78%(RSD=1.34%,n=9)。结论:该方法环保、简便、准确度和精密度良好,适用于测定注射用醋酸奥曲肽中乙酸的含量。
离子抑制色谱法;注射用醋酸奥曲肽;乙酸
醋酸奥曲肽(Octerotide Acetate)是人工合成的一种八肽环状化合物,为生长抑素的衍生物,可有效抑制5-羟色胺、胃泌素、血管活性肠肽、促胰液素等激素的分泌,临床多用于治疗胰腺炎、消化道出血等症[1-2]。合成醋酸奥曲肽的最后一步为将其制成乙酸盐,因此检测和控制醋酸奥曲肽中乙酸的含量对保证其质量具有重要的意义。多肽药物中乙酸的测定方法主要包括离子交换色谱法[3]、反相高效液相色谱法[4-8]、气相色谱法[9-10]。2015年版《中国药典》(四部)通则0872规定利用反相高效液相色谱法检测合成多肽中的乙酸[11],但在该方法中,乙酸出峰后还需耗费较长的时间于高比例有机相下洗脱多肽,以达到净化色谱柱的目的;且笔者也曾用此方法检测了醋酸奥曲肽中乙酸的含量,但由于乙酸含量较低,检测效果不佳。以醋酸曲普瑞林为代表的某些多肽药物,药典规定除反相高效液相色谱法外,还可以按照2015年版《中国药典》(二部)收载的离子交换色谱法来测定乙酸的含量[12]。但此方法对仪器的要求较高,检测器则通常采用电导检测器,通用性较差。气相色谱法则是测定有机溶剂残留的常见方法,但其常用的氢火焰离子化(FID)检测器对乙酸的响应度较其他有机溶剂低[4],且检测结果受气流稳定性的影响,重复性相对不佳。
离子抑制色谱法(IEC)常被用于复杂有机弱酸的分离检测[13],以及天然产物和药物中反离子的检测[14]。其经典理论认为,色谱柱填料为有一定交联度的全磺化氢离子(H+)型高容量离子交换树脂,高密度的电荷使得树脂颗粒间隙中的液体与树脂微孔中吸留的液体之间形成了一层半透膜,即唐楠膜。当强电离组分通过时,受到唐楠排斥作用而不能进入微孔,弱电离组分如乙酸则能够进入微孔得到保留[15]。本文采用IEC法测定注射用醋酸奥曲肽中乙酸的含量,以期为药典标准的完善提供参考。
1 材料
1.1仪器
Thermo U-3000型双三元液相色谱仪,包括双三元低压梯度泵、自动在线脱气机、自动进样器、柱温箱、可变波长检测器、变色龙7.2工作站(美国Thermo Fisher Scientific公司);Milli-Q Integral型超纯水器(德国Merck Millipore公司);XP205型十万分之一电子分析天平(瑞士Mettler-Toledo公司)。
1.2药品与试剂
注射用醋酸奥曲肽(上海第一生化药业有限公司,批号:1412801、130803Y、1410703,规格:0.1 mg/瓶);三水合乙酸钠对照品(德国Sigma公司,批号:WXBB4849V,纯度:99.0%);三氟乙酸、乙酸、甲酸为色谱纯,硫酸为分析纯,水为超纯水。
2 方法与结果
2.1色谱条件
色谱柱:Rezex ROA-Organic Acid H+(300 mm×7.8 mm,8 µm);流动相:0.002 5 mol/L硫酸溶液;流速:0.5 ml/min;检测波长:210 nm;柱温:45℃;进样量:100 μl。
2.2溶液的制备
2.2.1对照品溶液精密称取三水合乙酸钠对照品18.45 mg(相当于乙酸7.999 mg),置于200 ml量瓶中,加水溶解并稀释至刻度,摇匀,作为对照品贮备液。精密量取上述对照品贮备液5 ml,置于10 ml量瓶中,加水稀释至刻度,摇匀,经0.22µm微孔滤膜滤过,即得。
2.2.2供试品溶液取样品适量,加水溶解并稀释,制成每1 ml含醋酸奥曲肽0.1 mg的供试品溶液。
2.2.3阴性对照溶液以水作为本试验的阴性对照溶液。
2.3系统适用性试验
取“2.2”项下阴性对照溶液、对照品溶液和供试品溶液各100 μl,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。由图1可知,在该色谱条件下,各成分均能达到基线分离,分离度>3;理论板数以乙酸峰计为6 160;保留时间为19.34 min。结果表明,其他成分对测定无干扰。
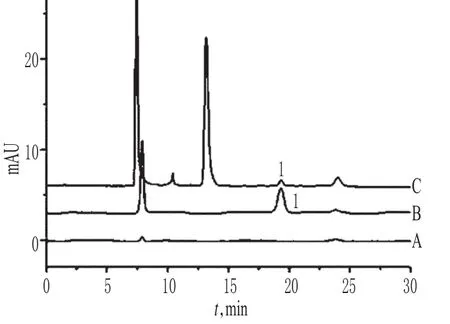
图1 离子抑制色谱图A.阴性对照;B.对照品;C.供试品;1.乙酸Fig 1 IEC chromatogramsA.negative control;B.reference substance;C.test sample;1.acetic acid
2.4专属性试验
取0.1%乙酸、0.1%甲酸及0.1%三氟乙酸溶液适量,混匀,作为专属性试验溶液。取上述溶液100 μl,按“2.1”项下色谱条件进样测定,记录色谱,详见图2。由图2可知,在该色谱条件下,三氟乙酸在死时间内被洗脱;甲酸与乙酸在色谱柱上保留情况良好,分离度为2.5。结果表明,常见有机酸基对乙酸的检测无影响。
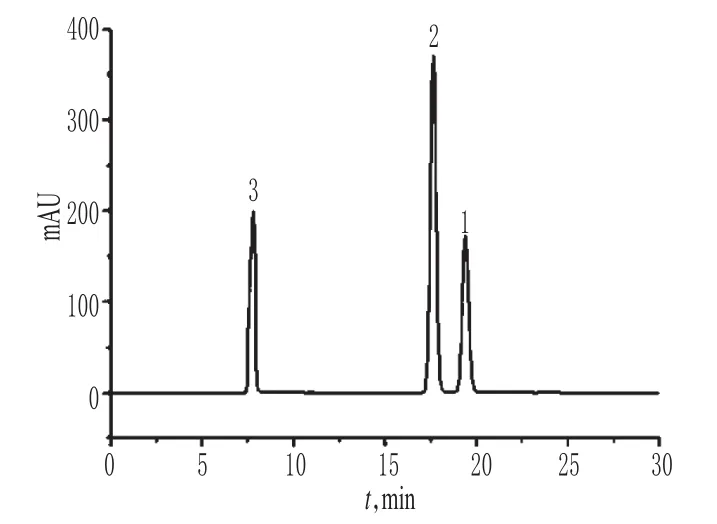
图2 离子抑制色谱图1.乙酸;2.甲酸;3.三氟乙酸Fig 2 IEC chromatograms1.acetic acid;2.formic acid;3.trifluoroacetic acid
2.5线性关系考察
精密称取三水合乙酸钠对照品适量,加水溶解制成乙酸质量浓度分别为0.394 4、0.788 9、1.972、7.889、19.72、39.45、78.89µg/ml的系列溶液。取上述系列溶液适量,按“2.1”项下色谱条件进样测定,记录峰面积。以质量浓度(x,µg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得乙酸的回归方程为y=0.111 5 x-0.002 8(r=0.999 9)。结果表明,乙酸的检测质量浓度线性范围为0.394 4~78.89µg/ml。
2.6定量限与检测限考察
取“2.2.1”项下对照品溶液适量,等倍逐步稀释,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。当信噪比为10∶1时,得定量限;当信噪比为3∶1时,得检测限。结果,乙酸的定量限为197.2 ng/ml;检测限为78.89 ng/ml。
2.7精密度试验
取“2.2.1”项下对照品溶液适量,按“2.1”项下色谱条件进样测定,记录峰面积。结果,乙酸峰面积的RSD=1.98%(n= 6),表明仪器精密度良好。
2.8稳定性试验
取“2.2”项下供试品溶液适量,分别于室温下放置0、2、4、6、8、10 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,乙酸峰面积的RSD=1.58%(n=6),表明供试品溶液在室温下10 h内稳定性良好。
2.9重复性试验
取样品(批号:1412801)适量,按“2.2.2”项下方法制备供试品溶液,共6份,再按“2.1”项下色谱条件进样测定,记录峰面积。结果,乙酸峰面积的RSD=1.34%(n=6),表明本方法重复性良好。
2.10加样回收率试验
取样品(批号:1412801)9瓶(每瓶含乙酸2.15 mg),分别转移至10 ml量瓶中,分别加入低、中、高质量的三水合乙酸钠对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表1。

表1 加样回收率试验结果(n=9)Tab 1 Result of recovery test(n=9)
2.11样品含量测定
取3批样品各适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按外标法计算乙酸的含量,结果见表2。

表2 样品含量测定结果(n=3,%)Tab 2 Result of content determination of samples(n=3,%)
3 讨论
3.1色谱柱的选择
IEC法使用的固定相主要是高交换容量的阳离子交换树脂。严晋婴等[16]探讨了IEC所用树脂的特性,其指出若想要加快唐楠膜的平衡来达到高效分离的目的,树脂应具有较大的孔径和恰当的交联度。而菲罗门公司生产的Rezex ROA-Organic Acid H+色谱柱的填料具有较大的孔径和约8%的交联度,非常适合分析以乙酸为代表的有机酸。因此,选择Rezex ROA-OrganicAcid H+为本试验的色谱柱。
3.2流动相的选择
有机酸在水溶液中的离解平衡受pH的影响。降低pH可抑制有机酸的解离,从而延长有机酸在色谱柱上的保留时间。理论上,凡是能提供氢离子(H+)的溶液都可作为IEC的淋洗液。淋洗液主要分为3类:水、有机酸与无机酸。在本试验中,笔者选择(稀)硫酸为唯一的流动相,没有使用任何有机溶剂,绿色环保;且多肽药物在(稀)硫酸创造的酸性条件下离子化而没有保留,乙酸则几乎不解离,保留情况良好。这简化了主成分的洗脱过程,并使得乙酸的检测不受干扰。因此,本试验选择(稀)硫酸为流动相。
3.3进样量的设定
本试验供试品溶液中乙酸的含量较低,预试验结果显示,参照常规的试验方法将进样量设为20或10µl时,乙酸峰面积过小。因此,为保证检测效果,特将进样量设为100µl。
3.4检测条件的选择
在选择检测器时,考虑到硫酸与实验室现有的电喷雾检测器、电导检测器均不是十分兼容,而经紫外-可见全波长扫描验证,乙酸的最大吸收波长在210 nm左右,而硫酸在210 nm波长附近几乎没有吸收,进一步结合2015年版《中国药典》(四部)[11]中收载的标准,本试验最终选择使用紫外检测器在210 nm波长下进行乙酸含量的检测,通用性较强。
3.5常见有机酸的影响
笔者考察了甲酸和三氟乙酸对乙酸检测的影响。三氟乙酸的酸性远强于乙酸,于死体积处出峰;甲酸的酸性与乙酸十分接近,在色谱柱上保留良好,且甲酸与乙酸的分离度可达到2.5以上。这说明在本试验测定条件下,常见有机酸基对乙酸的检测无影响。随着分析时间的增加,色谱柱存在一定的钝化现象,因此乙酸和甲酸的分离度可以作为该色谱系统的适用性条件,当其不符合要求时,考虑对色谱柱进行净化。
3.6加样回收率试验结果
在加样回收率试验中,考虑到样品中乙酸含量低,于是在制备溶液时是将9瓶注射用醋酸奥曲肽中的内容物分别全部转移至量瓶中;且由于该制剂中乙酸含量远低于原料药,检测难度较大,加样回收率结果偏高。本试验仅对多肽药物中乙酸的检测方法进行了探索性研究,加样回收率试验的设计与试验结果有待进一步地优化及衡量。
3.7含量测定结果
2015年版《中国药典》(二部)中[12]收载的醋酸奥曲肽原料药标准规定乙酸含量应为5.0%~12.0%,但对醋酸奥曲肽各制剂中乙酸的含量并没有提出明确的限度。在本试验中,注射用醋酸奥曲肽中的乙酸含量仅为2%左右,这可能与制剂中含有适量的辅料有关。
综上所述,本试验采用IEC法测定注射用醋酸奥曲肽中乙酸的含量,方法环保、简便、准确度和精密度好,适用于测定注射用醋酸奥曲肽中乙酸的含量。
[1]刘根元.醋酸奥曲肽治疗急性胰腺炎的临床价值分析[J].中国处方药,2015,13(6):86.
[2]戴静毅,禹蔚琴.醋酸奥曲肽与泮托拉唑联合治疗肝硬化上消化道出血的疗效观察[J].中国医药指南,2012,10(5):202.
[3]邓锋,林向华.离子色谱法测定合成多肽类药物中醋酸的含量[J].中国当代医药,2014,21(22):56.
[4]鞠祖芳.高效液相色谱梯度洗脱法测定胸腺五肽原料药中醋酸和三氟醋酸的含量[J].医学信息,2010(4):946.
[5]黄萍,杨秀丽,王洋.高效液相色谱法测定醋酸奥曲肽中醋酸的含量[J].中国药师,2009,12(6):763.
[6]赖涛,刘春霞,李静,等.高效液相色谱法测定生长抑素中醋酸的含量[J].昆明医学院学报,2008(4):186.
[7]邱淑慧.高效液相色谱法测定多抗原肽MAP4原料药中醋酸和三氟醋酸的含量[J].生物技术世界,2014(9):143.[8]高春,高恒莹,姚瑛,等.高效液相色谱法测定醋酸奥曲肽中醋酸含量[J].中国生化药物杂志,2004,25(4):237.
[9]孟静娟,单风平.气相色谱法测定液相合成蛋氨酸脑啡肽中杂质残留量[J].微生物学杂志,2008,28(3):106.
[10]周晖,王东凯.气相色谱法对自制生长抑素中醋酸含量的测定[J].中国医科大学学报,2004,33(3):280.
[11]国家药典委员会.中华人民共和国药典:四部[S].2015年版.北京:中国医药科技出版社,2015:110.
[12]国家药典委员会.中华人民共和国药典:二部[S].2015年版.北京:中国医药科技出版社,2015:1 533、1 545.
[13]刘瑞,聂黎行,王钢力,等.离子排斥色谱法测定生脉注射液中的有机酸[J].分析化学,2012,40(3):489.
[14]宗艳萍,卢静华,山广志,等.高效离子抑制色谱法和高效离子交换色谱法检测琥珀酸去甲文拉法新中琥珀酸的含量[J].色谱,2016,34(2):189.
[15]钟展环.离子排斥色谱的分离机理及应用研究[J].分析测试仪器通讯,1995,5(4):205.
[16]严晋婴,施荫玉.离子排斥色谱用于低分子羧酸分析的进展[J].色谱,1993,11(1):13.
(编辑:刘柳)
Content Determination ofAceticAcid in OctreotideAcetate for Injection by IEC
LI Jinghua1,LIU Guixia1,LI Panpan1,WANG Zhiliang2,YAO Jing3,LI Zhuorong1,SHAN Guangzhi1(1.Institute of Medicinal Biotechnology,Chinese Academy of Medical Sciences,Beijing 100050,China;2.Weifang People's Hospital,Shandong Weifang 261041,China;3.National Institutes for Food and Drug Control,Beijing 100050,China)
OBJECTIVE:To determine the content of acetic acid in Octreotide acetate for injection by IEC,and provide reference for the improvement of pharmacopoeia standards.METHODS:The column was Rezex ROA-Organic Acid H+with mobile phase of 0.002 5 mol/L sulfuric acid at a flow rate of 0.5 ml/min,the detection wavelength was 210 nm,column temperature was 45℃,and injection volume was 100µl.RESULTS:The linear range of acetic acid was 0.394 4µg/ml-78.89µg/ml(r=0.999 9);RSDs of precision,stability and reproducibility tests were all lower than 2%;the limit of quantification was 197.2 ng/ml,and limit of detection was 78.89 ng/ml;recovery was 104.71%-109.78%(RSD=1.34%,n=9).CONCLUSIONS:The method is environmental and simple with good accuracy and precision,and suitable for the content determination of acetic acid in Octreotide acetate for injection.
IEC;Octreotide acetate for injection;Acetic acid
R917
A
1001-0408(2016)27-3867-03
10.6039/j.issn.1001-0408.2016.27.41
*硕士研究生。研究方向:药物分析。电话:010-63021345。E-mail:lijinghua010005@163.com
副研究员,博士。研究方向:药物分析。E-mail:shanguangzhi@imb.pumc.edu.cn
(2016-04-01
2016-06-12)
猜你喜欢
中西医结合心脑血管病杂志(2022年12期)2022-07-07 10:24:24
云南化工(2021年11期)2022-01-12 06:06:20
中华养生保健(2020年2期)2020-11-16 00:49:20
广州化工(2020年8期)2020-05-19 06:23:56
云南中医学院学报(2015年1期)2015-07-31 18:10:45
实用中西医结合临床(2015年7期)2015-02-28 16:30:36
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:56
中国药业(2014年17期)2014-05-26 09:07:49
中国药业(2014年21期)2014-05-26 08:56:48
河南医学研究(2014年7期)2014-02-27 14:53:23
