
SN38前药及其新剂型研究进展Δ
2016-11-18 03:05:17邓彩赟蒋成君张晓敏浙江科技学院生物与化学工程学院杭州31003杭州普施康生物科技有限公司杭州31001
中国药房 2016年28期
邓彩赟,蒋成君,张晓敏,余 波(1.浙江科技学院生物与化学工程学院,杭州 31003;.杭州普施康生物科技有限公司,杭州 31001)
SN38前药及其新剂型研究进展Δ
邓彩赟1*,蒋成君1#,张晓敏2,余波2(1.浙江科技学院生物与化学工程学院,杭州310023;2.杭州普施康生物科技有限公司,杭州310021)
目的:为合成新结构的7-乙基-10-羟基喜树碱(SN38)前药及其新剂型研究提供参考。方法:以“SN38”“Polymer”“7-ethyl-10-hydroxycamptothecin”“7-乙基-10-羟基喜树碱”“前药”等为
,组合查询1998年1月-2015年6月在SciFinder、中国知网、万方、维普、浙江省高校数字图书馆中关于SN38前药及其新剂型的相关文献,对SN38的特点、结构改造、制剂研究、研究前景和方向进行综述。结果与结论:共查询到相关文献80余篇,其中有效文献38篇。SN38几乎不溶于水(11~38 μg/ml),不溶解在大多数药用溶剂和油中,因此直接制成液体制剂受到限制。大多数文献报道在SN38的C-10和C-20位置进行化学结构修饰,可以改善其溶解性。某些SN38前药的纳米制剂已经被研究用于改善给药至癌细胞和组织,因此可以利用高通透性和滞留(EPR)效应优先将药物引导至肿瘤组织。在改善SN38溶解性的同时,还应注意相关药物的毒副作用。
7-乙基-10-羟基喜树碱;前药;溶解性;研究进展
喜树碱(CPT)是一个具有五环骨架的药物,其组成为1个(3,4-β)喹啉基团(A、B环)、1个氮茚(C、D环)和1个α-羟基-δ-内酯环(E环),具有1个手性碳原子C-20。CPT是抑制DNA拓扑异构酶1(TOP1)成熟的抗肿瘤药[1]。20α-羟基以及内酯环(E)和吡啶酮组(D)是抑制拓扑异构酶活性所必需的基团[2]。在C-9和C-10适当取代的类似物大多数具有增强药效的作用[3]。在所有CPT类似物中,7-乙基-10-羟基喜树碱(SN38)作为最具代表的一个活性类似物,是一种半合成的天然抗癌生物碱喜树碱类似物。羟基在C-10位置,乙基在C-7位置,这有利于提高SN38在生理环境下的稳定性,与其他的CPT类似物相比可以提高其药效[2]。SN38目前已经作为一种有效的抗癌药物而备受关注,其利用疏解超螺旋DNA的双螺旋结构和促进DNA的复制来调节DNA的拓扑结构,进而抑制TOP的合成。其通过形成三元复合物与DNA和TOP1在酪氨酸存在下发挥其治疗作用[4],结果,DNA复制失败,导致细胞死亡。纳米颗粒具有长时间循环的优点,有能力通过高通透性和滞留(EPR)效应在肿瘤组织积累。使用低剂量的纳米制剂,通过EPR效应能达到更高疗效、减少副作用。
笔者以“SN38”“Polymer”“7-ethyl-10-hydroxycamptothecin”“7-乙基-10-羟基喜树碱”“前药”等为关键词,组合查询1998年1月-2015年6月在SciFinder、中国知网、万方、维普、浙江省高校数字图书馆中关于SN38前药及其新剂型的相关文献。结果,共查询到相关文献80余篇,其中有效文献38篇。现对SN38的特点、结构改造、制剂研究、研究前景和方向进行综述,以期为合成新颖结构的SN38前药及其新剂型研究提供参考。
1 SN38的特点
SN38的摩尔质量为392.4 g/mol,在25下,电离常数(pKa)为2.01,分配系数(lgP)为2.65[5],几乎不溶于水(11~38 μg/ ml),不溶解在大多数药用溶剂和油中[6],因此直接制成液体制剂受到限制。经过一系列的溶解测试发现,SN38能溶解在0.5%(m/m)的二甲基亚砜、甲酸和二乙二醇单乙基醚中[6];还能溶解在0.1 mol/L的氢氧化钠溶液中,这表明SN38的开环形式是水溶性的。对CPT进行研究,发现SN38的内酯环是可逆的水解。处于C-20手性碳原子上的α-羟基促进了内酯环(E)的水解,并且生理环境pH 7.4是最有利于打开内酯的条件;SN38的内酯环在pH≤4.5时稳定,在pH>9.0时则完全水解形成羧酸盐形式[5]。在pH为6.7时,这两种形式是平衡的。由于开环的SN38羧酸盐不具有治疗效果,所以在生理环境(pH 7.4)下药物是否稳定是决定SN38治疗效果的一个重要因素。SN38在pH 7.4、温度37的生理液中,同时存在内酯形式和开环的羧酸盐形式。内酯形式大多与血浆蛋白(主要是白蛋白)结合,并且主要分布于肝、肾和小肠中[7]。在肝脏中,SN38通过尿苷二磷酸葡萄糖醛酸基转移酶1A(UGT1A)代谢并转化成水溶性的非活性代谢物——SN38葡萄糖醛酸(SN38G)。SN38经历肝肠循环,在少数的血浆浓度-时间分布的报告中出现反弹峰。在肝脏中,膜转运蛋白从血浆进入肝细胞摄取SN38。SN38和SN38G通过三磷酸腺苷结合盒(ABC)转运至胆道,接着消除进入肠道[8]。在肠道内,通过菌群分泌的β-葡萄糖醛酸苷酶可将SN38G再生为SN38,过量的SN38存在于肠道内会引起晚期衰弱性腹泻[9-10]。一小部分SN38G也可以通过肾脏排泄消除。SN38从血液中消除半衰期为10~209h[11-12]。CPT(A)、CPT-11(B)、SN38内酯(C)、SN38羧酸酯(D)的结构式与相互关系见图1。

图1 CPT(A)、CPT-11(B)、SN38内酯(C)、SN38羧酸酯(D)的结构式与相互关系
鉴于SN38难以溶解,得到的药理数据仅是SN38的水溶性前药伊立替康(CPT-11)给药后的数据。CPT-11由羧酸酯酶主要在肝脏中转化为更多的活性代谢物SN38[13-14],药效为SN38的1/1 000~1/100。CPT-11静脉或口服给药后,CPT-11和SN38的内酯或羧酸盐形式都可检测到。但仅有少部分的CPT-11转化为SN38,并且在不同患者间差别很大。CPT-11或SN38治疗最常见的剂量限制性毒性(DLT)为迟发性腹泻和骨髓抑制;约35%的治疗患者出现3~4级腹泻,14%~47%出现3~4级中性粒细胞减少[3]。结果显示,在肝脏中SN38经过葡萄糖醛酸化的代谢程度直接影响腹泻程度。经过代谢转换为水溶性衍生物SN38G的速率取决于尿苷二磷酸葡糖苷酸转移酶1A1(UGT1A1)基因多态性,并且不同患者之间差别很大[8,15]。由于SN38代谢有限,使得UGT1A1*28等位基因具有使中性粒细胞减少的高风险[8]。目前已经报道了各种关于提高SN38药效的研究报告,通过提高其水溶性或者在药物溶剂中的溶解度,稳定其内酯结构,降低SN38相关疗法的DLT。SN38(A)在肝脏中通过UGT1A1介导代谢得到SN38G(B)的结构图见图2。

图2 SN38(A)在肝脏中通过UGT1A1介导代谢得到SN38G(B)的结构图
2 结构改造和制剂研究
SN38的前药通常在SN38 C-10和C-20的位置进行修饰,且部分前药已进入临床前或临床试验的不同阶段,SN38的前药可分为亲水性前药、亲脂性前药和两亲性前药。SN38前药的一般结构式见图3,进入临床阶段的SN38前药见表1。
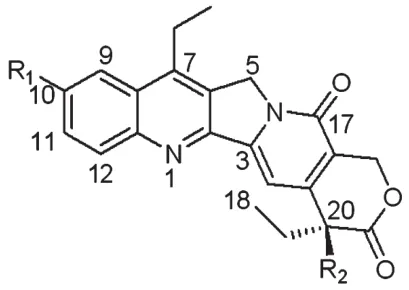
图3 SN38前药的一般结构式

表1 进入临床阶段的SN38前药
2.1亲水性SN38前药
为了能使SN38以溶液的形式给药,增加其溶解度是最常用的方法。一般可以选择可降解的水溶性高分子聚乙二醇(PEG)来修饰,修饰的位点为内酯环上C-20的羟基。PEG的修饰可避免药物被网状内皮系统吞噬,延长药物在体内的循环时间;PEG产生的空间位阻,可提高内酯环的稳定性;另外,PEG可提高药物的水溶性及肿瘤组织的被动聚集效应,增强药物的疗效[26]。CPT-11在体内经肝羧酸酯酶作用释放其活性药物SN38从而发挥抗肿瘤作用,但是由于CPT-11体内酶转化率低、个体间药动学性质不确定,导致其抗肿瘤活性存在较大的个体差异,又由于乙酰胆碱酯酶的抑制作用而引发毒副作用[27]。为了克服这些不足,专家设计了一系列亲水性SN38前药[27]:以线性氨基酸为连接子,氨基酸N端通过氨基甲酸酯键与SN38的C10相连,并选择生物相容性好的N-甲基哌嗪通过酰胺键与氨基酸分子C端相连,利用N-甲基哌嗪结构中氮原子的碱性与盐酸(HCl)成盐以提高水溶性。亲水性SN38前药一般结构式见图4。常见的亲水性SN38前药(a~k)见表2。

图4 亲水性SN38前药一般结构式

表2 常见的亲水性SN38前药(a~k)
CPT-11是通过肝羧酸酯酶介导的水解,将其转化为SN38;而DTS-108与这种限制性的治疗效果相反。DTS-108是高水溶性(>500 ng/ml)共轭物,它是SN38通过酯键与人的阳离子寡肽耦合得到的[28]。酯键允许在血浆中通过循环酯酶快速高效地释放,SN38的非肝特异性释放也有益于降低肝脏中SN38的葡萄糖醛酸。给犬iv DTS-108(20 mg/kg)或CPT-11(30 mg/kg)后,DTS-108浓度比CPT-11下降得更迅速;DTS-108给药后,SN38的药-时曲线下面积(AUC)比CPT-11高264倍。Ⅰ期临床试验显示,使用DTS-108治疗的患者,剂量限制腹泻消失了,而中性粒细胞减少和输液反应(瘙痒性荨麻疹)与DTS-108的DLT有关。DTS-108的最大耐受剂量(MTD)高达416 mg/m2,并且与CPT-11推荐剂量(RD)350 mg/m2相比,其RD为313 mg/m2[16]。此外,合成一系列C-10羟基位被含氮杂环取代的SN38衍生物,具有改进水溶解度的作用[21]。这些衍生物在体外显示与CPT-11具有相似的药效,但是需要在动物模型中进一步验证[29]。
大分子和纳米颗粒经过循环系统在肿瘤部位的积累是众所周知的现象,称为EPR效应[30]。EPR效应可被用于减少所需的总剂量,以实现等同的治疗效果,因此具有很大的潜力以克服与SN38治疗相关的当前毒性。SN38已经与亲水性聚合物如PEG化纳米石墨烯氧化物共价结合,以提高水的溶解度以及通过EPR效应提高肿瘤选择性[18]。
除了通过EPR效应来提高SN38的肿瘤选择性,癌细胞表面的特异性受体或配体也被用作靶向部分以增强SN38在肿瘤的积累。例如,癌胚抗原相关细胞黏附分子5(CEACAM5)在固体肿瘤表面过度表达;单克隆抗体(mAb)、拉贝珠单抗(hMN14)对CEACAM5有特定的选择性。几种双官能团SN38前药(即hMN14-CL2-SN38),也称为抗体-药物偶联物(ADC),其合成在C-20位置和短链PEG上利用了不同的交联剂[31]。在体外细胞结合以及其结合物的细胞毒性研究显示,游离的mAb以及其结合物比游离的SN38更具细胞毒性。Govindan SV等[32]在裸鼠人结肠癌(LS174T)和肺转移模型的临床前研究中发现,当使用hMN14-CL2-SN38或hMN14-CL2-SN38(Et)治疗时,相比于对照组和33倍高累积量的CPT-11,存活时间显著延长。对CEACAM5雌性动物使用hMN14-CL2-SN38是无效的,而对雄性动物却可以100%存活,这证明了药物对肿瘤细胞的特异性靶向作用。最近,Cardillo TM、Sharkey RM等[33-34]也评估了一些类似的结构,如SN38结合到滋养层细胞表面抗原2(Trop-2)特异性抗体、依帕珠单抗(Vmab,抗CD22)和维妥珠单抗(抗CD20),结果表明快速内化特性可增强药物的活性。ADC的成功取决于SN38和抗体以及该抗体对肿瘤的选择性之间的结合化学。但是,若要更透彻地了解SN38释放模式的影响、抗体内化的过程以及药物通过细胞膜的疗效和毒性,需要确定这些偶联物的有效剂量和安全剂量。mAb和SN38偶联物结构式见图5。
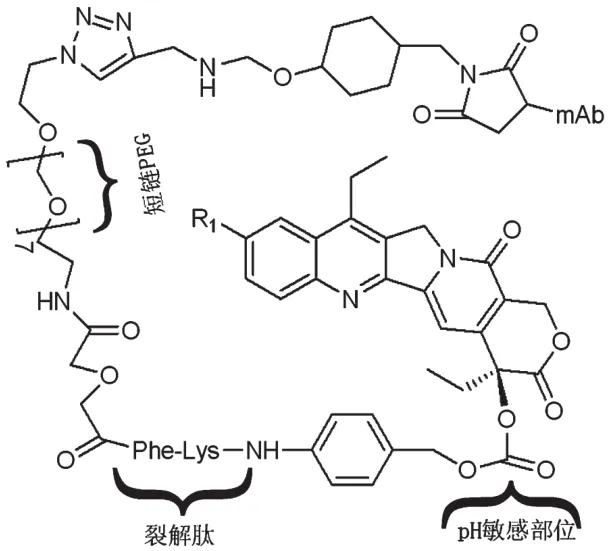
图5 mAb和SN38偶联物结构式
在肿瘤和利用酶促活性的环境下,提出了两种靶向治疗的方式。例如,肿瘤相关的血管过度表达整合素αvβ3,它可以被包含精氨酸-甘氨酸-天冬氨酸(RGD)的环状五肽所识别[20],这种识别是通过吲哚醌的结构与SN38相连。吲哚醌在心肌黄酶(DTD)的存在下会裂解,允许酶定向生物还原药物传送至某些升高的DTD水平肿瘤细胞。对人宫颈癌KB细胞系列的初步研究表明,在生物还原条件下约50%~70%会受到增长的抑制,而前药在缺乏DTD的情况下,无毒颗粒粒径可达300 nm。
第二种报道中利用糖苷酶(如β-D-葡萄糖醛酸酶)在肿瘤细胞中的丰度。其通常存在于非常低浓度的血液循环中,并且主要可以在肿瘤中从SN38切割葡萄糖醛酸基团[19]。为了证明这个概念,合成了SN38G。对HT-29癌细胞的细胞毒性研究表明,SN38G与母体药物相比,在缺失β-D-葡萄糖醛酸酶的情况下,毒性降低了70倍,而药效在β-D-葡萄糖醛酸酶的存在下完全恢复。这表明相比于SN38,其增加了肿瘤特异性。进一步的研究需要了解这种前体药物的有效性和毒性以及合适的给药方式,如通过细菌菌群在肠道分泌β-D-葡萄糖醛酸酶可在肠道内将SN38G转化为SN38。
2.2亲脂性SN38前药
通过前体药物制剂增加SN38的脂溶性,具有改善脂质制剂载药量、水解后维持内酯形式、提高母体药物通过细胞膜渗透性等优点。
例如,Lundberg BB[23]通过在SN38的C10位置结合油酸合成了SN38-油酸。相比于SN38,SN38-油酸在三酰甘油中的溶解度提高了3.6倍;SN38-油酸在脂质体或脂质乳剂中比SN38高出5~7.5倍的掺入率。在脂质体或脂质乳剂中进行抗T-47D和Caco-2细胞系的细胞活力研究表明,SN38-油酸半数抑制浓度(IC50)值约为1~3 μmol/L,这类似于SN38在二甲基亚砜-乙醇溶液中给药,证实了细胞毒性的保护。荧光显微镜观察结果表明,脂质体的SN38-油酸积累在细胞间,而游离的SN38积累在细胞内。由于使用亲脂性前药会增强肿瘤细胞的吸收,因此对多重抗药性的癌症是有益的。Val通过酯键连接到SN38 C-20位置(图6B)生成Val-SN38[24]。Val-SN38改善了细胞内的积累,其在MCF-7细胞的积累量是SN38的5倍,从而不会引起显著的多药耐药性[35]。目前认为是通过各种氨基酸转运增强摄取而改善Val-SN38的积累。但是前药的稳定性差并且容易在磷酸盐缓冲溶液(PBS溶液)、MCF-7细胞系和血浆中转化为非活性羧酸盐。因此,前药的进一步优化,以更好地控制将其转化为羧酸盐形式的转化率,可得到更有效的前药;另外,前药可以掺入脂质制剂,以稳定其内酯形式。SN38-油酸(A)、Val-SN38(B)的结构式见图6。
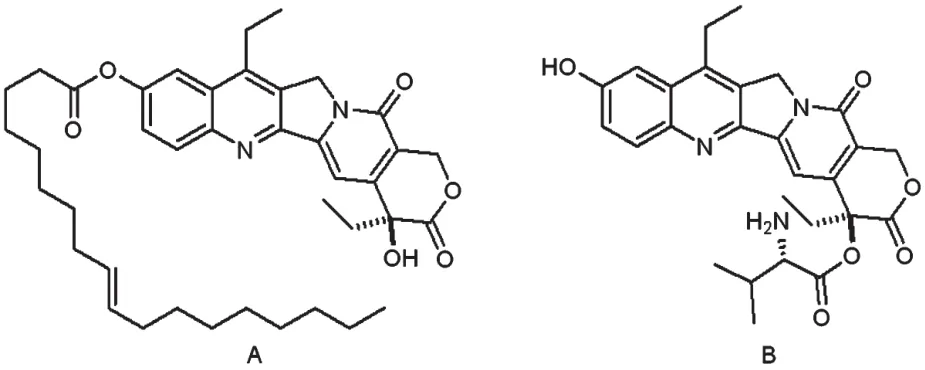
图6 SN38-油酸(A)、Val-SN38(B)的结构式
SN2310是一种SN38的新型亲脂性前药,是由α-生育酚与SN38通过琥珀酸盐连接在C-10位置上酯化而成[15,36]。SN2310能在油中提高它的溶解性,允许可注射油包水纳米乳液制剂以10 mg/ml掺入SN2310。Marier JF等[15]在对小鼠、大鼠、犬和猴进行SN2310乳液的临床前研究中发现,与CPT-11相比,改善曝光和延长SN38的消除半衰期,可以减少其剂量限制性腹泻。25 mg/m2SN2310注射乳剂的MTD约为CPT-11(750 mg/m2)MTD的3%。25 mg/m2剂量的SN2310乳剂相比于125 mg/m2剂量的CPT-11治疗,具有类似的暴露水平。此外,SN2310乳剂的平均cmax和t1/2分别比CPT-11高3.6倍和20倍。SN38的浓度峰值出现在SN2310乳剂静脉滴注结束时,这表明SN2310在血浆和肝脏中快速转化为SN38。但是,目前这些研究还不确定的是,在SN2310注射后延长SN38的暴露时间,是跟乳剂有关,还是与SN38以亲脂性前药的形式给药有关。亲脂性SN38前药SN2310的结构式见图7。

图7 亲脂性SN38前药SN2310的结构式
2.3两亲性SN38前药
两亲性SN38前药由于其两亲性质,能在水溶液环境中形成胶束。在水溶液中将SN38与亲水性聚合物结合以形成胶束,或者利用胶束载体递送SN38,这两种方法都用来增加水溶性,在pH诱导和/或酶水解情况下保护药物,在生理环境下延长药物循环时间,并且通过EPR效应增加在肿瘤部位的积累[37]。
将小分子质量的OEG结合在SN38的C20位置,以形成类似表面活性剂的两亲性前药——OEG-SN38[25]。干燥的OEGSN38中SN38的载药量为36%(m/m),并且水化后胶束粒径为28 nm。该制剂在PBS溶液中稳定存在35 h(4.71%释放)。OEG-SN38的细胞毒性是CPT-11的11~200倍。在体内,对Bcap37人乳腺癌异种移植的裸鼠给药OEG-SN38,与相同剂量(相当于10 mg/kg的SN38)的CPT-11表现出类似的肿瘤生长抑制效果。另外,也可以在C-10位的酚羟基上引入酯键,并同时引入OEG,从而获得具有两亲性结构的SN38前药分子,后者在水中能形成外层为OEG、内层为SN38的纳米药物,从而避免了SN38与水分子直接接触。
NK012代表另一种SN38胶束形成前药。SN38与PEG和聚谷氨酸(PGlu)的两亲性嵌段共聚物中的PGlu结合,形成PEG-PGlu(SN38),其中谷氨酸与SN38通过苯基酯键结合[7]。前体药物在水溶液中组装成胶束(NK012),其平均粒径为20 nm。冻干的NK012制剂含有20%(m/m)的SN38,在5%的葡萄糖溶液中稳定存在48 h(97%)。PEG-PGlu(SN38)聚合物的苯基酯键在血浆(存在或不存在水解酶)中水解并释放出SN38[11]。对HT-29人大肠癌异种移植的小鼠进行体内研究显示,当使用NK012(30 mg/kg)时,血浆中SN38的AUC比使用CPT-11(66.7 mg/kg)高14倍,这可能与NK012具有更长的循环时间和EPR效应,并且可以维持SN38在血液中的有效浓度有关[11]。对小鼠肝脏转移140 d的体内研究表明,使用NK012(30 mg/kg)、CPT-11(66.7 mg/kg)和对照组的存活率分别为100%、0、0[38],这证明了在小鼠模型中NK012的有效性和安全性。
3 结语
SN38代表最活跃的CPT类似物。由于各种化学和药理学的限制,在市场上,它只能以CPT-11注射剂的形式获得。虽然CPT-11完全批准用于各种癌症的治疗,但是只有一小部分的注射CPT-11转化为活性的母体药物SN38,同时关于CPT-11的各种副作用也有报道。因此,有必要改善SN38的化学结构来达到更有效、更安全的治疗效果。目前的前药尝试克服了SN38溶解度差的缺点,涉及到用高度水溶性基团对C-10进行改性,以提高它的水溶性;或者用脂溶性基团,使其能进入基于脂质的载体;同时,对C-20进行改性,以稳定SN38的内酯环。进一步的研究可以对SN38进行结构修饰,改善其溶解性,将其制成纳米制剂等,增强其肿瘤靶向性。在提高药效的同时,还应注意降低毒副作用,这将有助于更好地发挥SN38的抗肿瘤作用。
[1]胡文晋,胡巍,方芸.喜树碱类药物抗肿瘤机制研究进展[J].中国药房,2012,23(27):2581.
[2]Ulukan H,Swaan PW.Camptothecins:a review of their chemotherapeutic potential[J].Drugs,2002,62(14):2039.
[3]Garcia-Carbonero R,Supko JG.Current perspectives on the clinical experience,pharmacology,and continued development of the camptothecins[J].Clin Cancer Res,2002,8(3):641.
[4]Pommier Y.DNA topoisomeraseⅠinhibitors:chemistry,biology and interfacial inhibition[J].Chem Rev,2009,109(7):2894.
[5]Thakur R,Sivakumar B,Savva M.Thermodynamic studies and loading of 7-ethyl-10-hydroxycamptothecin into mesoporous silica particles MCM-41 in strongly acidic solutions[J].J Phys Chem B,2010,114(17):5903.
[6]Roger E,Lagarce F,Benoit JP.Development and characterization of a novel lipid nanocapsule formulation of SN38 for oral administration[J].European Journal of Pharmaceutics&Biopharmaceutics,2011,79(1):181.
[7]Koizumi F,Kitagawa M,Negishi T,et al.Novel SN-38-incorporating polymeric micelles,NK012,eradicate vascular endothelial growth factor-secreting bulky tumors[J]. Cancer Res,2006,66(20):10048.
[8]Innocenti F,Kroetz DL,Schuetz E,et al.Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics[J].J Clin Oncol,2009,27(16):2604.
[9]Takakura A,Kurita A,Asahara T,et al.Rapid deconjugation of SN-38 glucuronide and adsorption of released free SN-38 by intestinal microorganisms in rat[J].Oncol Lett,2012,3(3):520.
[10]Fujiwara Y,Minami H.An overview of the recent progress in irinotecan pharmacogenetics[J].Pharmacogenomics,2010,11(3):391.
[11]Hamaguchi T,Doi T,Eguchi-Nakajima T,et al.PhaseⅠstudy of NK012,a novel SN-38-incorporating micellarnanoparticle,in adult patients with solid tumors[J].Clin Cancer Res,2010,16(20):5058.
[12]Kurzrock R,Goel S,Wheler J,et al.Safety,pharmacokinetics,and activity of EZN-2208,a novel conjugate of polyethylene glycol and SN38,in patients with advanced malignancies[J].Cancer,2012,118(24):6144.
[13]Schoemaker NE,Kuppens IELM,Huinink WWTB,et al. PhaseⅠstudy of an oral formulation of irinotecan administered daily for 14 days every 3 weeks in patients with advanced solid tumours[J].Cancer Chemother Pharmacol,2005,55(3):263.
[14]Pitot HC,Adjei AA,Reid JM,et al.A phaseⅠand pharmacokinetic study of a powder-filled capsule formulation of oral irinotecan(CPT-11)given daily for 5 days every 3 weeks in patients with advanced solid tumors[J].Cancer Chemother Pharmacol,2006,58(2):165.
[15]Marier JF,Pheng L,Trinh MM,et al.Pharmacokinetics of SN2310,an injectable emulsion that incorporates a new derivative of SN-38 in patients with advanced solid tumors [J].J Pharm Sci,2011,100(10):4536.
[16]Coriat R,Faivre SJ,Dreyer C,et al.First-in-human phaseⅠ and pharmacokinetic study of DTS-108 in patients with advanced carcinomas[J].J Clin Oncol,2012,30(15):2557.
[17]Kolhatkar RB,Swaan P,Ghandehari H.Potential oral delivery of 7-ethyl-10-hydroxy-camptothecin(SN-38)using poly(amidoamine)dendrimers[J].Pharm Res,2008,25(7):1723.
[18]Liu Z,Robinson JT,Sun X,et al.PEGylated nanographene oxide for delivery of water-insoluble cancer drugs [J].J Am Chem Soc,2008,130(33):10876.
[19]Angenault S,Thirot S,Schmidt F,et al.Cancer chemotherapy:a SN-38(7-ethyl-10-hydroxycamptothecin)glucuronide prodrug for treatment by a PMT(Prodrug Mono Therapy)strategy[J].Bioorg Med Chem Lett,2003,13(5):947.
[20]Huang B,Desai A,Tang S,et al.The synthesis of a c(RGDyK)targeted SN38 prodrug with an indolequinone structure for bioreductive drug release[J].Org Lett,2010,12(7):1384.
[21]Li QY,Deng XQ,Zu YG,et al.Cytotoxicity and topoⅠtargeting activity of substituted 10:nitrogenous heterocyclic aromatic group derivatives of SN-38[J].European Journal of Medicinal Chemistry,2010,45(7):3200.
[22]Williams CC,Thang SH,Hantke T,et al.RAFT-derived polymer-drug conjugates:poly(hydroxypropyl methacrylamide)(HPMA)-7-ethyl-10-hydroxycamptothecin(SN-38)conjugates[J].Chem Med Chem,2012,7(2):281.
[23]Lundberg BB.Biologically active camptothecin derivatives for incorporation into liposome bilayers and lipid emulsions[J].Anti-cancer Drug Design,1998,13(5):453.
[24]Kwak EY,Shim WS,Chang JE,et al.Enhanced intracellular accumulation of a non-nucleoside anti-cancer agent via increased uptake of its valine ester prodrug through amino acid transporters[J].Xenobiotica,2012,42(7):603.
[25]Zhang H,Wang J,Mao W,et al.Novel SN38 conjugateforming nanoparticles as anticancer prodrug:in vitro and in vivo studies[J].J Control Release,2013,166(2):147.
[26]徐蓓华,周惠燕.喜树碱-聚乙二醇前药的合成及体内外释药特性[J].中国药科大学学报,2012,43(2):142.
[27] 周沫,何新华,史卫国,等.10-羟基喜树碱前药的合成及其初步活性评价[J].中国药物化学杂志,2014,24(5):337.
[28]Meyer-Losic F,Nicolazzi C,Quinonero J,et al.DTS-108,a novel peptidic prodrug of SN38:in vivo efficacy and toxicokinetic studies[J].Clin Cancer Res,2008,14(7):2145.
[29]Li Q,Zu Y,Shi R,et al.Synthesis and antitumor activity of novel 10-substituted camptothecin analogues[J].Bioorg Med Chem,2006,14(21):7175.
[30]Jaracz S,Chen J,Kuznetsova LV,et al.Recent advances in tumor-targeting anticancer drug conjugates[J].Bioorg Med Chem,2005,13(17):5043.
[31]Moon SJ,Govindan SV,Cardillo TM,et al.Antibody conjugates of 7-ethyl-10-hydroxycamptothecin(SN-38)for targeted cancer chemotherapy[J].J Med Chem,2008,51(21):6916.
[32]Govindan SV,Cardillo TM,Moon SJ,et al.CEACAM5-targeted therapy of human colonic and pancreatic cancer xenografts with potent labetuzumab-SN-38 immunoconjugates[J].Clin Cancer Res,2009,15(19):6052.
[33]Cardillo TM,Govindan SV,Sharkey RM,et al.Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers:preclinical studies in human cancer xenograft models and monkeys[J].Clin Cancer Res,2011,17(10):3157.
[34]Sharkey RM,Govindan SV,Cardillo TM,et al.Epratuzumab-SN-38:a new antibody-drug conjugate for the therapy of hematologic malignancies[J].Mol Cancer Ther,2012,11(1):224.
[35]Kwak EY,Choi MK,Yang SG,et al.Investigation into the efficacy of Val-SN-38,a valine-ester prodrug of the anti-cancer agent SN-38[J].Biomol Ther:Seoul,2012,20(3):326.
[36]Zhang Y,Gold LC.Tocopherol-modified therapeutic drug compounds:US,7223770B2[P].2007-05-29.
[37]Gong J,Chen M,Zheng Y,et al.Polymeric micelles drug delivery system in oncology[J].J Control Release,2012,159(3):312.
[38]Guo M,Rong WT,Hou J,et al.Mechanisms of chitosancoated poly(lactic-co-glycolic acid)nanoparticles for improving oral absorption of 7-ethyl-10-hydroxycamptothecin[J].Nanotechnology,2013,24(24):245101.
(编辑:余庆华)
R944
A
1001-0408(2016)28-4005-05
10.6039/j.issn.1001-0408.2016.28.36
浙江省自然科学基金资助项目(No.LY15B060008)
*硕士研究生。研究方向:药物制剂。电话:0571-85070383。E-mail:2272446801@qq.com
高级工程师,博士。研究方向:药物制剂。电话:0571-85070383。E-mail:jcj312@163.com
(2015-12-31
2016-04-20)
猜你喜欢
食品与生物技术学报(2022年1期)2023-01-11 09:11:11
盐科学与化工(2020年9期)2020-09-23 02:41:58
宁夏医学杂志(2020年4期)2020-03-01 13:16:20
宁夏医学杂志(2020年3期)2020-02-27 14:17:11
新生代(2019年8期)2019-10-28 06:39:30
中成药(2018年2期)2018-05-09 07:20:05
分析化学(2017年12期)2017-12-25 12:03:53
中学生数理化·高三版(2016年12期)2017-03-02 19:21:37
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
中国卫生标准管理(2015年15期)2015-01-26 20:32:38
