
位置功能候选基因HMGA1、C6orf106和ENSSSCG00000023160与猪肢蹄结实度的关联性
2016-11-17 08:16:40张徐非候利娟邱恒清黄路生郭源梅
中国农业科学 2016年20期
张徐非,候利娟,邱恒清,黄路生,郭源梅
(1温州医科大学动物实验中心,浙江温州 325000;2江西农业大学种猪遗传改良与养殖技术国家重点实验室,南昌 330045)
位置功能候选基因HMGA1、C6orf106和ENSSSCG00000023160与猪肢蹄结实度的关联性
张徐非1,2,候利娟2,邱恒清2,黄路生2,郭源梅2
(1温州医科大学动物实验中心,浙江温州 325000;2江西农业大学种猪遗传改良与养殖技术国家重点实验室,南昌 330045)
【目的】建立一种通过内在的四肢骨关节评分来评估猪肢蹄结实度的方法,并计算关节评分与表观评估的肢蹄结实度之间的相关系数。此外,在F2、莱芜、二花脸、苏太和杜长大5个猪群中,研究三个位置功能候选基因HMGA1、C6orf106和ENSSSCG00000023160与猪肢蹄结实度之间的关联性。【方法】根据关节面裂痕的大小和深浅以及损伤的严重程度,对5块四肢骨的关节进行评分(1—5分)。如果关节面裂痕很大且很深,或损伤很严重,则评为1分;如果关节面没有裂痕和损伤,则评为5分。评分越高,说明关节越健康,肢蹄越结实。此外,基于笔者前期全基因组关联分析的结果,在猪7号染色体最强关联SNP两侧翼各0.2 Mb的区域内,筛选出HMGA1、C6orf106和ENSSSCG00000023160三个位置功能候选基因。在F2群体中,通过基因测序,搜寻这3个基因的多态位点,并根据多态位点在6个物种间的保守性,筛选出11个多态位点。利用Taqman探针,对3个HMGA1位点和3个C6orf106位点进行基因分型,而另一个基因的5个多态位点则通过基因型填补(genotype imputation)方法进行基因分型。最后,利用R软件GenABEL程序包,分析最小等位基因频率(MAF)大于0.05的多态位点与肢蹄结实度之间的关联性。【结果】在C6orf106和ENSSSCG00000023160基因中,分别鉴别到174和5个多态位点。关节评分之间呈显著的正相关,绝大部关节评分与蹄趾、肢蹄和步态评分之间无相关,而与肱二头肌长度和重量呈显著的负相关。公猪的肩胛骨关节评分极显著低于母猪的评分,但是臂骨肩关节和后肢跗关节评分显著高于母猪相应的关节评分。在F2群体中,3个候选基因均与肢蹄结实度关联,但是ENSSSCG00000023160的关联程度不如另外2个基因强,可以排除它为肢蹄结实度的因果基因。因此,该基因没有在其余的4个群体中进行检测。在二花脸群体中,HMGA1的g.2029C>T和g.3155A>G位点均与肢蹄结实度关联,另一个位点的MAF小于0.05。在其它3个群体中,HMGA1 3个位点的MAF都小于0.05。在莱芜群体中,仅C6orf106的g.6953T>C位点的MAF大于0.05,该位点与肢蹄结实度显著关联。在苏太和杜长大群体中,C6orf106的g.2054T>C和g.6953T>C位点的MAF大于0.05,但它们与肢蹄结实度性状之间无显著关联。【结论】建立了一套利用四肢骨关节评分来评估肢蹄结实度的方法,该方法是对现有的表观评估方法的重要补充。因为蹄趾、肢蹄和步态评分与关节评分无相关,所以它们不能取代关节评分。关联分析结果排除了ENSSSCG00000023160是肢蹄结实度因果基因,但没有排除HMGA1和C6orf106的可能性,因此,有必要对这2个基因进行更深入的研究。
猪;肢蹄结实度;关节评分;关联分析;基因型填补
0 引言
【研究意义】肢蹄结实度反映猪前后肢蹄的结实程度,一般通过外在的肢蹄评分和步态评分来度量[1]。不结实的肢蹄是一种病,称为肢蹄软弱(leg weakness)。猪肢蹄软弱发病率高,病因复杂,且难以治愈,导致大量种猪和生产母猪被动淘汰。据报道:在通过了生产性能测定的后备公猪中,有20%—50%的后备公猪因肢蹄软弱而被淘汰[2-3];在生产母猪中,也有6.1%—15%的母猪因肢蹄软弱而被淘汰[4]。肢蹄结实度作为一个复杂性状,它受遗传和环境因素的共同影响。猪肢蹄结实度具有中等遗传力,其遗传力为0.1—0.5[3,5-7]。ROTHSCHILD等在一个杜洛克群体中对肢蹄结实度进行5个世代的歧化选择后,正向选择群体的肢蹄结实度显著高于反向选择群体[7]。因此,对肢蹄结实度的遗传解析可以为它的遗传改良奠定基础。【前人研究进展】利用全基组连锁分析,定位了很多肢蹄结实度的数量性状基因座(quantitative trait locus, QTL);通过全基因组关联分析(genomewide association analysis,GWAS),鉴别了一些与肢蹄结实度关联的单核苷多态(single nucleotide polymorphism,SNP)。在猪的QTL数据库中,一共收录了278个控制肢蹄结实度的QTL或与之关联的SNP[8]。本研究小组在白色杜洛克×二花脸的F2资源家系中,利用183个微卫星标记,通过全基因组扫描,一共定位到42个影响肢蹄结实度的QTL,其中7号染色体SW1856—S0102区间内的QTL效应最大、置信区间最小(只有6 cM)[9]。利用Illumina公司猪60K SNP芯片,在该F2资源群体和苏太群体中,通过GWAS,将影响前后肢步态评分的基因位点定位在7号染色体上一个2.15 Mb的连锁不平衡框内,其最强关联的SNP为35.18 Mb的MARC0033464[10]。【本研究切入点】目前,对肢蹄结实度的测定仅停留在外观上,缺乏一种内在的评估方法。另外,7号染色体最强关联区域内的基因与肢蹄结实之间度的关联也没有报道。【拟解决的关键问题】建立一种通过内在的关节评分来度量肢蹄结实度的方法,并与现有方法作比较,评估该方法的必要性。另外,在7号染色体最强关联区域内筛选3个位置功能候选基因,并对其多态位点进行搜寻,然后在每个候选基因中筛选3个保守的多态位点,在F2、苏太、莱芜、二花脸和杜长大5个群体中进行关联分析,寻找与肢蹄结实度显著关联的多态位点,为肢蹄结实度的标记辅助选择奠定基础。
1 材料与方法
1.1 试验材料
1.1.1 试验群体 本研究一共用了5个试验猪群,即F2资源群体、苏太猪、二花脸、莱芜猪和杜长大群体。F2资源群体以2头白色杜洛克公猪和17头二花脸母猪为亲本,杂交产生F2代[11]。苏太猪是杜洛克公猪和太湖母猪杂交,经19个世代对繁殖和生长性状的选育,培育而成的母系品种[10]。331头二花脸和314头莱芜猪分别购自江苏省焦西二花脸合作社和山东省莱芜原种猪场。在4—5月龄从购买猪场运至江西省南昌市国鸿生态园猪场进行肥育。二花脸公母猪均进行了阉割,莱芜猪只对公猪进行阉割。这4个群体在肥育期间均饲喂含3 100 kJ可消化能、16%粗蛋白和0.78%赖氨酸的全价配合饲料,自由采食和饮水,且采用相同的饲养管理方式,自由采食和饮水。F2资源群体和苏太猪在240日龄左右屠宰,二花脸和莱芜猪在300日龄左右屠宰。610头杜长大来自江西国鸿集团修水商品猪场。公猪在出生时阉割,母猪不阉割。分阶段饲喂相应的配合饲料,自由采食和饮水,体重在110 kg左右屠宰。
1.1.2 表型测定 各个表型的测定,分别由同一人根据相同的标准在江西省南昌市进行测定。肢蹄评分和步态评分采用GUO等建立的方法在饲养场地进行测定[9]。在F2资源群体(2002—2006年)和苏太猪(2010—2011年)和莱芜猪(2012—2014年)群体中,肢蹄评分和步态评分都在220日龄左右进行测定,莱芜猪(2012—2014年)群体在300日龄左右进行评定。屠宰后,前后蹄从屠体上分割下来,根据蹄子的大小、均匀度、受损程度等进行评分。蹄子大、均匀且没有损伤评5分;蹄子很小、非常不均匀或损伤严重评1分。左侧的肱二头肌完整地从前肢剥离下来,用电子天平和游标卡尺分别测定其重量和长度。前后肢四肢骨的主要关节面,根据表1的评分标准进行评分。
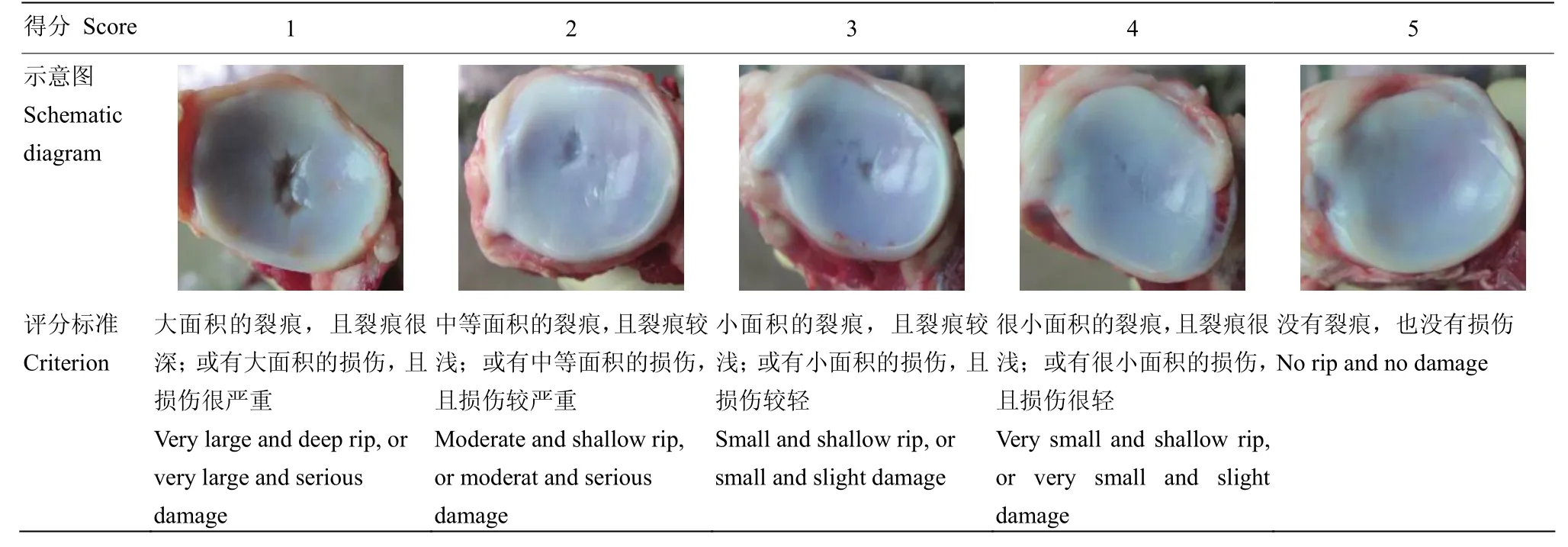
表1 猪关节面评分标准Table1 The criteria for joint surface score
1.1.3 基因DNA的提取 个体屠宰后,用剪刀剪取适量的耳组织或脾脏,贮存于装有75%的酒精溶液的EP管中备用。采用常规酚氯仿法从耳组织或脾脏中提取基因组DNA,经质量检测和浓度测定后稀释成50 ng·μL-1的工作液备用。
1.2 试验方法
1.2.1 位置功能候选基因的筛选及其SNP的搜寻在7号染色体上,以最强关联SNP为中心的0.4 Mb范围内一共有6个基因。HMGA1编码一种影响软骨细胞生长和分化的非组蛋白[12];C6orf06基因可能与骨骼生长相关,因为在C6orf06和HMGA1区域内鉴别到与人类身高显著关联的SNP[13-15];ENSSSCG00000023160的功能未知;这3个基因可能会影响肢蹄结实度,因此把它们作为肢蹄结实度的候选基因。RPS10编码一种核糖体蛋白S10,与人的先天性纯红细胞再生障碍性贫血关联[16];SPDEF编码上皮特异性ETs转录因子,与小肠杯状细胞的分化和成熟有关[17];PACSIN1编码神经元蛋白激酶C酪蛋白激酶底物1,调节神经元轴突的延伸和分支[18];这3个基因的功能均与肢蹄结实度不相关,因此可以排除。
从Ensembl网站上(http://www.ensembl.org/index. html)获取猪HMGA1、C6orf106和ENSSSCG00000023160 3个候选基因的基因组序列。根据其基因组序列,使用在线引物设计软件Primer3(http://frodo.wi.mit.edu/)分别设计16、50和5对引物对这3个基因的多态位点进行搜寻。
HMGA1和C6orf106 SNP基因多态位点搜寻的模板为3头F1公猪(耳号分别为17、29和41号)组成的DNA池。ENSSSCG00000023160则对所有的F0代个体单独进行测序,搜寻多态位点。PCR扩增后产物用琼脂糖凝胶电泳检测PCR扩增的有效性,合格的DNA样品经QIAquick DNA纯化试剂盒纯化后,送上海生工进行双向测序,获得所需序列。用DNAStar软件包的Seqman程序进行序列分析,鉴别多态位点。
1.2.2 多态位点的筛选及基因型判定 用Clustal W软件,选取猪、狗、奶牛、人、鼠和兔子6种哺乳动物DNA序列,对C6orf106进行保守性分析[19]。选取3个在这6种动物间保守且具有代表性的SNP用于基因判型,并利用相应限制性内切酶进行酶切验证其多态性。设计3对探针和引物,使用7900HT Fast Real-time PCR System分别对g.2054T>C、g.6953T>C和g34542A>T进行基因分型[20]。
沈虎群对HMGA1进行测序,搜寻到9个SNP,并在F2资源群体中对其中的g.1149C>T、g.2029C>T和g.3155A>G进行基因型检测[21]。在F2资源群体中,本研究直接使用他的分型结果。在另外的4个群体中,对这3个位点进行基因分型。
ENSSSCG00000023160基因采用基因型填补法(genotype imputation)来获取F2个体所有多态位点的基因型[22]。首先对全部的F0代和F1代个体的测序,获取F0和F1代个体该基因所有多态位点的基因型。然后在该基因两侧翼临近区域内选取15个多态性好的60K SNP芯片上的SNP位点(F0、F1和F2个体的基因型都已知),与该基因的基因型进行整合(F2代个体的基因型均为缺失),利用SimWalk2.9软件,构建所有个体的单倍型[23]。最后根据个体的单倍型来重建 F2个体的基因型。为了评估基因型填补法的准确性,在60K SNP芯片中,选取5个位于该基因两侧翼、且F0和F1代个体的基因型分别与该基因5个多态位点相类似的SNP,保留F0和F1代个体的基因型,把F2个体的基因型设为缺失,通过上述方法获取F2个体的基因。芯片检测的基因型和基因填补获得的基因型之间吻合度可以用来评估基因填补的准确性。吻合度越高,基因型填补法获取的基因型就越准确。
1.3 统计分析方法
1.3.1 表型相关及性别差异检验 表型简单统计量的计算及性别差异检验都在统计软件SAS9.0(SAS Institute Inc., Cary, NC, USA)上完成的。CORR过程用于计算表型之间的简单相关系数;TTEST过程检验表型在性别之间的差异。
1.3.2 关联分析 利用R软件中的GenABEL软件包,使用下面模型对肢蹄结实度相关性状进行关联分析[24]:

其中,y是表型向量;b是固定效应向量,包括性别和批次,分析肱二头肌长度和重量是还包括胴体重;u是加性遗传效应向量,服从N(0, G σ2α),G为基因组亲缘关系矩阵,利用猪60K芯片常染色体上SNP的计算得到[25-26];σ2α为加性方差;a为SNP等位基因的替代效应;X和Z分别为b和u的指示矩阵;s是a的指示向量;e是残差向量,服从N(0, Iσ2e)。
2 结果
2.1 多态位点搜寻
设计了50对引物对C6orf106进行全基因多态位点的搜寻,一共搜寻到多态位点个SNP[20]。用Clustal W软件对这174个多态位点进行保守性分析,有9个SNP在猪、狗、奶牛、人、鼠和兔子中是保守的[20]。从这9个保守的SNP中,选取3个具有代表的SNP,即g.2054T>C、g.6953T>C和g34542A>T,在这5个群体进行基因分型。
设计了5对引物对ENSSSCG00000023160进行全基因多态位点的搜寻,一共搜寻得到5个多态位点,即g.1129G>C、g.2284C>G、g.2430C>T、g.2813G>A和g.3231AA>--。利用基因填补法获取F2资源群体中F2个体的基因型。根据两侧翼5个SNP的基因填补的模拟结果,基因填补的错误率分别为2.73%、0、0.2%、1.0%和0.4%。

表2 性状之间的表型相关系数Table2 The phenotypic correlation coefficients among the measured traits
2.2 多态位点判型
在资源家系F2群体中,3个候选基因的11个多态位点的最小等位基因频率(MAF)均大于0.3。在其余4个群体中,只对C6orf106和HMGA1的6个多态位点进行了分型。在苏太和杜长大群体中,只有C6orf106前2个位点的MAF大于0.05;在二花脸群体中,只有HMGA1后2个位点的MAF大于0.05;在莱芜猪群体中,只有C6orf106第2个位点的MAF大于0.05。MAF大于0.05的位点用于后续的关联分析。
2.3 表型相关分析
在莱芜、二花脸和杜长大混合群体中,表型之间的简单相关系数见表2。肱二头肌长度和重量与前后蹄和前后肢肢蹄评分之间呈极显著正相关,而与关节评分之间呈显著负相关(除肱二头肌重量和前臂骨腕关节评分不显著之外)。肱二头肌长度与前肢步态评分呈显著负相关,与后肢步态评分不相关。肱二头肌重量与前肢步态评分不相关,与后肢步态评分呈显著正相关。前蹄评分除了与肱二头肌长度和重量以及后蹄评分呈正相关外,还与臂骨肩关节、前臂骨肘关节、前臂骨腕关节和股骨髋关节评分呈负相关,与其他性状不相关。后蹄评分除了与肱二头肌长度和重量以及前蹄评分呈正相关外,还与肩胛骨关节和臂骨肘关节评分呈显著的负相关,与肢蹄评分和步态评分呈显著正相关,与其他性状不相关。关节评分之间呈显著正相关(除肩胛骨关节与前臂骨肘关节评分以及前臂骨腕关节与肩胛骨关节、臂骨肘关节和小腿骨跗关节评分之间无相关外),与肢蹄和步态评分之间无相关(除股骨膝关节评分与前肢肢蹄和步态评分呈正相关、前臂骨腕关节评分与后肢肢蹄和步态评分呈负相关以及臂骨肩关节评分和前肢步态评分呈正相关外)。肢蹄与步态之间呈极显著的正相关。后肢跗关节评分只在杜长大群体中进行了测量,它与前蹄评分(r= 0.0172,P= 0.6726)和后蹄评分(r= 0.0612,P= 0.1322)之间无相关。
2.4 性别差异检验
性别对表型的影响见表3。公猪的肩胛骨关节评分极显著低于母猪的肩胛骨关节评分,但是臂骨肩关节和后肢跗关节评分显著高于母猪的相应关节的评分,其余性状在性别之间没有显著差异。

表3 不同性别对肢蹄结实度的影响Table3 The effects of sexes on the measured traits
2.5 关联分析结果
在F2资源群体中,11个多态位点与肢蹄结实度关联分析结果见表4。HMGA1基因的g.2029C>T和g.3155A>G位点与肱二头肌长度和重量、肢蹄评分和步态评分6个性状均显著关联,而g.1149C>T位点与肢蹄评分和前肢步态评分显著关联,与另外3个性状不关联。C6orf106基因的g.34542A>T位点与6个性状均显著关联,而另外2个位点与步态评分和前肢肢蹄评分显著关联,与另外3个性状不关联。ENSSSCG00000023160基因不与肱二头肌重量关联。在剩下的5个性状中,g.2430C>T和g.3231AA>--位点与它们显著关联,g.2284C>G和g.1129G>C位点分别与除前肢肢蹄评分和肱二头肌长度之外的4个性状显著关联,而另一个位点不与任何性状关联。在这11个多态位点中,HMGA1的g.3155A>G与前肢步态评分的关联程度最强。

表4 F2资源群体中11个多态位点与肢蹄结实度关联分析Table4 The association analysis results between 11 loci and leg soundness in the F2population
在其余4个群体中,HMGA1和C6orf106与肢蹄结实度关联分析结果见表5。在莱芜群体中,只有C6orf106的g.6953T>C位点的MAF大于0.05。该位点与臂骨肩关节、股骨膝关节、后肢步态和前臂骨肘关节评分显著关联,与其余性状之间不关联。在二花脸群体中,只有HMGA1的g.2029C>T和g.3155A>G位点的MAF大于0.05。前者与股骨髋关节评分极显著关联,后者与股骨髋关节和臂骨肘关节评分极显著关联,与其余性状不关联。在苏太和杜长大群体中,只有C6orf106的g.2054T>C和g.6953T>C位点的MAF大于0.05,但它们均与肢蹄结实度性状之间不关联。
3 讨论
本研究一共使用了5个试验猪群。选择了F2群体是因为7号染色体上的QTL是在F2资源群体中鉴别到的。因此,只有在这群体中显著关联的位点,才有可能是因果突变位点。苏太猪与F2资源群体有相似的来源,他们的祖代都是杜洛克和二花脸。但是苏太猪经历了18个世代的重组,连锁不平衡区域会显著的小于F2群体,因此选择这个群体有利于区分因果位点和连锁不平衡位点。二花脸是F2资源群体的祖代之一,使用这个品种有利于了解这个QTL的起源。莱芜猪是中国著名地方品种之一,对莱芜猪肢蹄结实度的研究,有利于该地方品种的开发和利用。杜长大是目前中国商品猪生产的主要杂交模式,在杜长大群体中开展肢蹄结实度的研究,可以为商品猪肢蹄结实度的分子育种奠定基础。
群体对肢蹄结实度的影响是极显著的(数据没有展示)。杜长大群体的蹄趾评分显著高于两个地方品种,二花脸的前蹄趾评分比莱芜猪的要高,但是后蹄趾评分比莱芜猪要低。苏太猪的前后肢的肢蹄评分和步态评分均极显著高于莱芜猪。总体来说二花脸的关节评分比莱芜猪要高,如肩胛骨关节评分、臂骨肘关节评分、股骨膝关节评分和小腿骨跗关节评分,但莱芜猪的前臂骨腕关节评分比二花脸的高。
DRAPER等报道在肢蹄软弱品系中,肱二头肌的长度和重量显著大于肢蹄正常和结实的品系[27]。在F2群体中,前肢步态评分与肱二头肌的长度和重量呈显著的负相关,而肢蹄评分与它们呈显著正相关[9]。在苏太群体中,肱二头肌的长度和重量与步态和肢蹄评分不相关[10]。在莱芜、二花脸和杜长大混合群体中,前肢步态评分与肱二头肌长度呈显著的负相关,这重复了前人的结果。外观的蹄趾、肢蹄和步态评分与关节评分之间不相关,因此,它们不能替代关节评分。

表5 HMGA1和C6orf106 的4个SNP与肢蹄结实度关联分析Table5 The association analysis results between 4 SNPs of HMGA1 and C6orf106 and leg soundness
在F2群体和苏太群体中,母猪的肢蹄结实度高于公猪[10]。在莱芜、二花脸和杜长大混合群体中,公猪的肢蹄和步态评分以及肱二头肌长度显著小于母猪的,但是都没有达到显著水平,这和前人的结果相似[10]。
在F2群体中,ENSSSCG00000023160 的g.2284C>G、g.2430C>T和g.3231AA>--与前肢步态评分相关联,但关联显著水平不如HMGA1的两个位点(表4)。其余位点在不同群体中与肢蹄软弱的表型性状关联性不强,未达到统计学显著水平。虽然基因填补存在一定的错误,导致检测效率的下降,但是模拟结果显示错误在3%以下(与前人的结果类似[22]),对关联分析的结果影响不会很大。基于上述结果,可以排除该基因为肢蹄结实度的因果基因。因此,在其余的4个群体中,没有必要对该基因进行相关的研究。
HMGA1能提高软骨细胞增殖活性[12],并通过调控透明软骨细胞增殖和分化来影响软骨组织的修复[28]。在关节炎患者体内,HMGA1和IGFBP-3蛋白表达量均上调[29]。在F2和二花脸群体中,该基因均与肢蹄结实度相关性状显著关联,在其他3个群体中,该的3个体位点的MAF小于0.05,没有足够的检测效率,因此没有进行关联分析。因此,它仍有可能是肢蹄结实度因果基因。
在F2群体和莱芜群体中,C6orf106与肢体结实度性状显著地关联。在二花脸群体中,3个SNP的MAF均小于0.05。在苏太和杜长大群体中,g.34542A>T位点的MAF小于0.05,另外2个SNP的MAF虽然大于0.05,但是它们与已测的肢蹄结实度不关联。考虑到这个基因还有100多个多态位点没有检测,因此,不能排除其为因果基因的可能性。
4 结论
本研究建立了一种通过内在的关节评分来评估肢蹄结实度的方法。相关分析结果表明外观的蹄趾、肢蹄和步态评分与关节评分之间无显著相关性,因此,关节评分是对现有肢蹄结实度度量方法的重要补充。性别对肢蹄结实度有显著的影响,在进行肢蹄结实度的遗传解析中,需要考虑性别效应。关联分析结果排除了ENSSSCG00000023160是肢蹄结实度因果基因的可能性,但是HMGA1和C6orf106的可能性没有排除,有必要对这两个基因进行更深入的研究。
[1] 候利娟, 张徐非, 郭源梅. 猪肢蹄结实度的遗传解析进展. 猪业科学, 2013(12): 94-97.
HOU L J, ZHANG X F, GUO Y M. The advance of genetic deciphering of leg soundness in pigs. Swine Industry Science,2013(12): 94-97. (in Chinese)
[2] STEENBERGEN E J VAN. Description and evaluation of a linears coring system for exteriortraits in pigs. Livestock Production Science,1989, 23: 163-181.
[3] WEBB A J, RUSSELL W S, SALES D I. Genetics of leg weakness in performance-tested boars. Animal Production, 1983, 36: 117-130.
[4] STALDER K, SERENIUS T. Sow longevity scrutinized. National Hog Farmer, 2004, 49(7): 26-30.
[5] BERESKIN B. Genetic aspects of feet and leg soundness in swine. Journal of Animal Science, 1979, 48: 1322-1328.
[6] JØRGENSEN B, ANDERSEN S. Genetic parameters for osteochondrosis in Danish Landrace and Yorkshire boars and correlations with leg weakness and production traits. Animal Science, 2000, 71: 427-434.
[7] ROTHSCHILD M F, CHRISTIAN L L. Genetic control of front-leg weakness in Duroc swine. I. Direct response to five generations of divergent selection. Livestock Production Science, 1988, 19: 459-471.
[8] HU Z L, PARK C A, WU X L, REECY J M. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Research, 2013,41(Database issue): 871-879.
[9] GUO Y M, AI H S, REN J, WANG G J, WEN Y, MAO H R, LAN L T,MA J W, BRENIG B, ROTHSCHILD M F. A whole genome scan for quantitative trait loci for leg weakness and its related traits in a large F2 intercross population between White Duroc and Erhualian. Journal of Animal Science, 2009, 87(5): 1569-1575.
[10] GUO Y M, ZHANG X F, REN J, AI H S, MA J W, HUANG L S. A joint genomewide association analysis of pig leg weakness and its related traits in an F2 population and a Sutai population. Journal of Animal Science, 2013, 91(9): 4060-4068.
[11] GUO Y, MAO H, REN J, YAN X, DUAN Y, YANG G, REN D,ZHANG Z, YANG B, OUYANG J. A linkage map of the porcine genome from a large-scale White Duroc x Erhualian resource population and evaluation of factors affecting recombination rates. Animal Genetics, 2009, 40(1): 47-52.
[12] CLEYNEN I, VAN DE VEN W J. The HMGA proteins: a myriad of functions (Review). International Journal of Oncology, 2008, 32(2): 289-305.
[13] WEEDON M N, LANGO H, LINDGREN C M, WALLACE C,EVANS D M, MANGINO M, FREATHY R M, PERRY J R,STEVENS S, HALL A S. Genome-wide association analysis identifies 20 loci that influence adult height. Nature Genetics, 2008, 40(5): 575-583.
[14] BERNDT S I, GUSTAFSSON S, MAGI R, GANNA A, WHEELER E,FEITOSA M F, JUSTICE A E, MONDA K L, CROTEAU-CHONKA D C, DAY F R. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nature Genetics, 2013, 45(5): 501-512.
[15] SORANZO N, RIVADENEIRA F, CHINAPPEN-HORSLEY U,MALKINA I, RICHARDS J B, HAMMOND N, STOLK L, NICA A,INOUYE M, HOFMAN A. Meta-analysis of genome-wide scans for human adult stature identifies novel Loci and associations with measures of skeletal frame size. PLoS Genetics, 2009, 5(4): e1000445.
[16] DOHERTY L, SHEEN M R, VLACHOS A, CHOESMEL V,O'DONOHUE M F, CLINTON C, SCHNEIDER H E, SIEFF C A,NEWBURGER P E, BALL S E. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. American Journal of Human Genetics, 2010, 86(2): 222-228.
[17] NOAH T K, KAZANJIAN A, WHITSETT J, SHROYER N F. SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Experimental Cell Research,2010, 316(3): 452-465.
[18] LIU Y, LÜ K, LI Z, YU A C, CHEN J, TENG J. PACSIN1, a Tau-interacting protein, regulates axonal elongation and branching by facilitating microtubule instability. Journal of Biological Chemistry,2012, 287(47): 39911-39924.
[19] LARKIN M A, BLACKSHIELDS G, BROWN N P, CHENNA R,MCGETTIGAN P A, MCWILLIAM H, VALENTIN F, WALLACE I M, WILM A, LOPEZ R. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England), 2007, 23(21):2947-2948.
[20] 张徐非. 位置候选基因HMGA1、C6orf106和ENSSSCG00000023160与猪肢蹄结实度的关联性研究[D]. 江西南昌: 江西农业大学硕士学位论文, 2013.
ZHANG X F. Evaluation of effects of positional candidate genes HMGA1, C6orf106 and ENSSSCG00000023160 on pig leg soundness traits[D]. Jiangxi, Nanchang: Jiangxi Agricultural University, 2013.(in Chinese)
[21] 沈虎群. 位置候选基因HMGA1、SRPK1、ZNF76与猪四肢骨骼长度的相关性研究[D]. 南昌: 江西农业大学, 2009.
Shen H Q. Evaluation of effects of positional candidate genes HMGA1,SRPK1 and ZNF76 in the SSC7 QTL region on pig limb bone lengths[D]. Nanchang: Jiangxi Agricultural University, 2009. (in Chinese)
[22] Burdick J T, Chen W M, Abecasis G R, Cheung V G. In silico method for inferring genotypes in pedigrees. Nature Genetics, 2006, 38(9): 1002-1004.
[23] Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. American Journal of Human Genetics, 1996, 58(6): 1323-1337.
[24] Aulchenko Y S, Ripke S, Isaacs A, van Duijn C M. GenABEL: an R library for genome-wide association analysis. Bioinformatics (Oxford,England), 2007, 23(10): 1294-1296.
[25] Amin N, van Duijn C M, Aulchenko Y S. A genomic background based method for association analysis in related individuals. PLoS ONE, 2007, 2(12): e1274.
[26] Astle W, Balding D J. Population structure and cryptic relatedness in genetic association studies. Statistical Science, 2009, 24(4): 451-471.
[27] Draper D D, Rothschild M F, Christian L L. Effects of divergent selection for leg weakness on muscle and bone characteristics in Duroc swine. Genetics Selection Evolution, 1992, 24: 363-374.
[28] Richter A, Hauschild G, Murua Escorbar H, Nolte I, Bullerdiek J. Application of high-mobility-group-A proteins increases the proliferative activity of chondrocytes in vitro. Tissue Engeering Part A, 2009, 15(3): 473-477.
[29] Gasparini G, De Gori M, Paonessa F, Chiefari E, Brunetti A, Galasso O. Functional relationship between high mobility group A1 (HMGA1)protein and insulin-like growth factor-binding protein 3 (IGFBP-3) in human chondrocytes. Arthritis Research and Therapy, 2012, 14(5): R207.
(责任编辑 林鉴非)
An Association Study of Positional and Functional Candidate Genes HMGA1, C6orf106 and ENSSSCG00000023160 with Leg Soundness in Pigs
ZHANG Xu-fei1,2, HOU Li-juan2, QIU Heng-qing2, HUANG Lu-sheng2, GUO Yuan-mei2
(1Laboratory Animal Center, Wenzhou Medical University, Wenzhou 325000, Zhejiang;2State Key Laboratory for Pig Genetic Improvement and Production Technology, Jiangxi Agricultural University, Nanchang 330045)
【Objective】 The objective of this study is to develop a method to access the leg soundness through scoring the joint of limb bone and calculate the simple correlation coefficients among the scores for leg soundness in pigs. Furthermore, the association between three positional and functional candidate genes, namely HMGA1, C6orf106 and ENSSSCG0000023160, and leg soundness was also studied in F2, Laiwu, Erhualian, Sutai and DLY populations.【Method】The joint of five limb bones were scored according to the size and depth of rip on the joint surface and the worn-out degree of the joint. If the rip on the joint surface is very big and deep or the joint is seriously worn out, the joint is scored 1. On the other hand, if there is no rip on the joint surface and the joint doesn’t have any degree of worn-out, the joint is scored 5. Higher the joint score is, healthier the joint is and sounder the leg is. Based on the authors’ previous genome-wide association studies, three genes HMGA1, C6orf106 and ENSSSCG0000023160 were screened as positional and functional candidate genes to leg soundness in a 0.4 Mb region centered on the top SNP on SSC7. To search the polymorphic loci of the three genes in the F2population, their DNA sequences were determined by a short-gun DNA sequence method. A total of 11 polymorphic loci were picked out according to their conservations among 6 species. The genotypes of 3 loci for HMGA1 and 3 loci for C6orf106 were determined using the Taqman method, and the genotypes of the other 5 loci for the third gene were inferred by genotype imputation just in the F2population. At last, the GenABEL package of R was used to perform the association analysis between the loci with MAF>0.05 and the traits.【Result】A total of 174 and 5 polymorphic loci were identified in C6orf106 and ENSSSCG00000023160 genes, respectively. Joint scores were positively correlated with each other and were negatively correlated with the length and weight of biceps brachii, but most of them had no correlation with toe, leg and gait scores. The male’s scapula joint score was significantly lower than the female’s, but arm shoulder joint score and focile hock joint score were significantly higher than the female’s corresponding joint score. In the F2population, all of the three genes were associated with leg soundness, but ENSSSCG00000023160 was weaker than the other two genes, therefore it was excluded as a candidate gene to leg soundness and was not genotyped in the other 4 populations. In the Erhualian population, two loci g.2029C>T and g.3155A>G of HMGA1 were significantly associated with leg soundness, and the other SNP lacked the polymorphism. In the other 3 populations, all of the 3 SNPs of HMGA1 were deficiently polymorphic. Only the g.6953T>C locus of C6orf106 had enough polymorphic in the Laiwu population, and it was associated with leg soundness. In the Sutai and DLY populations, only two loci g.2054T>C and g.6953T>C of C6orf106 were polymorphic, but none was associated with leg soundness.【Conclusion】A method of accessing the leg soundness has been proposed by scoring the joint of limb bones in pigs, and it is a crucial supplement method to access the leg soundness. Because toe, leg and gait scores are not correlation with the joint scores, they can’t replace the joint scores. The association analysis results excluded the ENSSSCG00000023160 gene as candidate gene to leg soundness, but both HMGA1 and C6orf106 genes were not excluded. Therefore, the two genes are worthy for further investigations.
pig; leg soundness; joint score; association analysis; genotype imputation
2015-08-27;接受日期:2016-08-05
国家自然科学基金(31060153,31460590)、江西省自然科学基金(20142BAB204017)
联系方式:张徐非,E-mail:604120811@qq.com。候利娟,E-mail:397999166@qq.com。张徐非和候利娟为同等贡献作者。通信作者郭源梅,Tel/Fax:0791-83813080;E-mail:gyuanmei@hotmail.com
猜你喜欢
科学大众(2024年5期)2024-03-06 09:40:34
系统工程学报(2021年4期)2021-12-21 06:21:08
新世纪智能(高一语文)(2020年12期)2020-06-01 08:14:20
阅读(快乐英语高年级)(2019年8期)2019-09-10 07:22:44
电子制作(2018年18期)2018-11-14 01:48:04
自动化学报(2018年6期)2018-07-23 02:55:42
小火炬·智漫悦读(2018年12期)2018-03-13 09:24:52
莫愁(2017年36期)2017-12-25 05:52:36
中国医药导报(2015年27期)2015-02-28 22:08:01
发明与创新(2015年33期)2015-02-27 10:40:00
