
GAP/环氧化合物体系胶片的制备与性能表征
2016-11-03 03:21郑启龙周伟良肖乐勤菅晓霞
固体火箭技术 2016年4期
郑启龙,周伟良,肖乐勤,菅晓霞
(南京理工大学 化工学院,南京 210094)
GAP/环氧化合物体系胶片的制备与性能表征
郑启龙,周伟良,肖乐勤,菅晓霞
(南京理工大学 化工学院,南京210094)
为探索一种新型非异氰酸酯固化体系,以端羟基聚叠氮缩水甘油醚(GAP)为研究对象,三羟甲基丙烷三缩水甘油醚(TMPTGE)为固化剂,通过实验筛选出六亚甲基四胺(HA)为固化催化剂,对GAP/TMPTGE/HA固化体系进行了研究。通过拉伸试验、DMA试验,研究了固化参数R和固化时间对GAP/TMPTGE胶片力学性能的影响,借助非等温DSC法,研究了GAP/TMPTGE/HA体系的固化动力学特征,并通过TG实验对胶片热性能进行了表征。结果表明,随着固化参数R的增大,胶片的断裂伸长率先增加后降低,拉伸强度不断增大;R=3.0时,胶片断裂伸长率达到最大值98%,此时拉伸强度为0.67 MPa,玻璃化转变温度为-34.8 ℃;胶片热分解分为2个阶段,对应的分解峰温分别为250 ℃和350 ℃。
聚叠氮基缩水甘油醚;环氧化合物;力学性能;热性能
0 引言
现行复合推进剂多为聚氨酯型推进剂,采用端羟基粘合剂和异氰酸酯固化体系[1],具有力学性能优良、使用温度范围广的特点。由于异氰酸酯基(—NCO)具有很高的极性和活泼性,易与水快速反应形成脲,并生成CO2,在推进剂中易形成气泡,影响其力学性能和燃烧性能[2]。因此,固化体系对环境湿度和推进剂组分含水量要求很高,使得一些性能优良但易吸湿的氧化剂(如硝酸铵、二硝酰胺铵等)难以应用于该体系中。另一方面,异氰酸酯反应生成的氨基甲酸酯基(—NH—COO—)耐水性有限,在一定条件下可发生水解,使得制品的抗渗透性、耐酸碱、耐候性降低[3]。聚叠氮基缩水甘油醚(GAP)推进剂作为一种高能、钝感、低特征信号的新型复合推进剂[4-5],也面临上述问题。有学者利用点击反应原理,采用炔基类化合物对叠氮粘合剂进行固化,固化反应机理为炔基和叠氮基之间1,3-偶极环加成生成三唑环,可避免固化体系对水分敏感的问题[6],但由于反应位置不确定,因而推进剂交联网络结构不可控。
GAP的端羟基在一定条件下可与环氧基团进行开环加成反应,以环氧化合物作为固化剂,同样可避免体系对水分敏感的问题。由于GAP中的羟基为仲羟基,在叔胺等碱性化合物存在下,其与环氧基团反应活性高于伯羟基和水,能够在较低温度下快速反应[7],因而环氧化合物固化GAP在工艺上具有可行性。
为探索一种新型非异氰酸酯固化体系,本文通过实验筛选出六亚甲基四胺(HA)为固化催化剂,研究了在HA催化下,固化参数、固化时间等对GAP/环氧化合物固化胶片力学性能的影响,同时还研究了环氧化合物固化GAP胶片的固化行为和热性能。
1 实验
1.1主要原料
GAP:相对分子质量3 430,羟值33.58 mgKOH/g,航天科技集团公司四十二所;三羟甲基丙烷三缩水甘油醚(TMPTGE):化学纯,嘉兴思诚化工有限公司;三氯化铝(AlCl3):分析纯;三氟化硼乙醚络合物(BF3·OEt2):分析纯;三乙胺(TEA):分析纯;六亚甲基四胺(HA):分析纯,国药集团化学试剂有限公司。
1.2试样制备
按配比将GAP与环氧固化剂TMPTGE混合,加入催化剂,80 ℃下搅拌2 h以保证混合均匀,真空除气泡,浇入聚四氟乙烯培养皿内,70 ℃烘箱内固化成胶片。
1.3性能测试
(1)傅立叶变换红外光谱(FTIR):采用德国Bruker光谱仪器公司的Tensor 27型傅立叶红外光谱仪进行衰减全反射红外测试,分辨率为4 cm-1,扫描次数为20次。
(2)固化行为测定:将固化体系混合均匀后,采用NETZSCH-STA 409型DSC差示扫描量热仪,进行升温速率分别为5、10、15、20 K/min的动态扫描,温度范围为50~180 ℃,测试环境为30 ml/min的N2气氛。
(3)力学性能测试:按照GBT 528—1998将胶片裁剪成标准哑铃形后,用美国英斯特朗公司的INSTRON 3367型精密万能材料试验机进行拉伸测试,测试温度为(20±2)℃,拉伸速率为100 mm/min,每个试样做3组平行实验求平均值。
(4)动态热机械性能测试:采用美国TA公司的DMA Q800 V7.0 Build 113测试样品的损耗模量、储能模量及损耗正切,温度范围为-70~50 ℃,测试频率1 Hz,振幅5 μm,升温速度为3 ℃/min。
2 结果及讨论
2.1不同催化剂催化GAP与TMPTGE的固化反应
不添加催化剂时,仅将GAP和TMPTGE进行混合固化,设置固化参数R值(即环氧基团与GAP中羟基的摩尔比值)分别为1.0、2.0、3.0、4.0,在70 ℃下固化1周,均无明显变化;时间延长至2周,仍为流动状态,不能固化。GAP中羟基可与环氧基发生开环加成反应,但醇羟基亲电性较弱,此温度下反应较难进行。因此,GAP与TMPTGE固化反应需要催化剂进行催化。常用的催化剂主要有路易斯酸催化剂和路易斯碱催化剂2种,前者主要包括三氯化铝、三氟化硼络合物等,后者主要包括无机碱、叔胺化合物等。为筛选GAP与TMPTGE固化反应的催化剂,在固化体系中分别加入三氯化铝(AlCl3)、三氟化硼乙醚络合物(BF3·OEt2)、三乙胺(TEA)和六亚甲基四胺(HA)进行固化实验,结果如表1所示。表1中,1~8#体系R=1.0,9#体系R=2.0;催化剂含量均为GAP质量的2%。
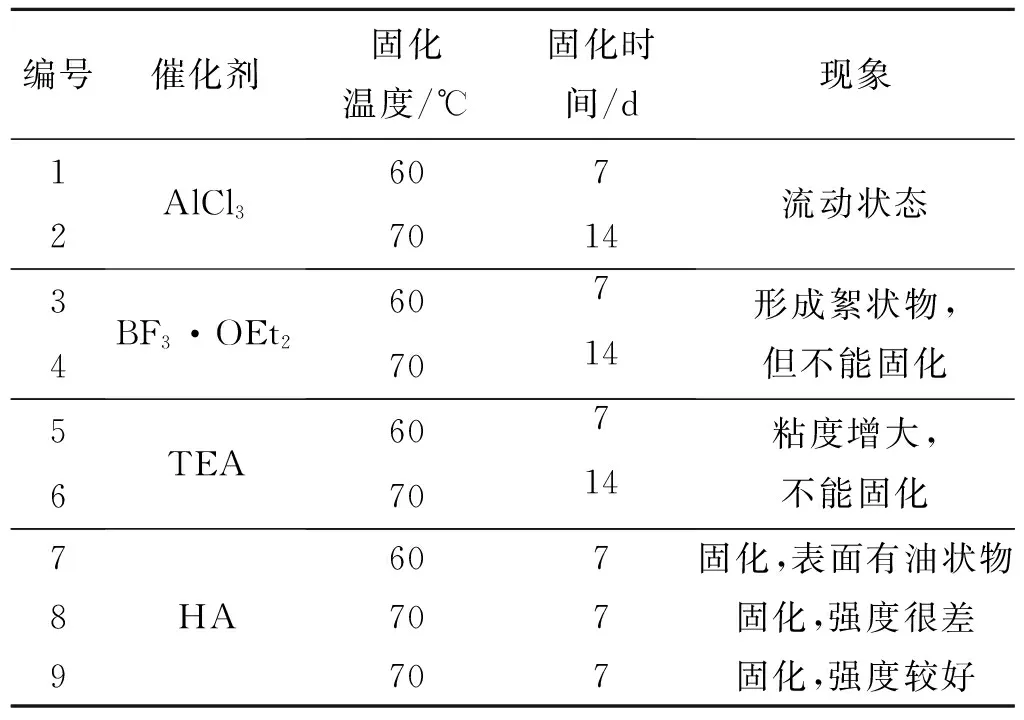
表1 不同催化剂的催化效果
通过表1可看出,AlCl3没有明显催化效果;BF3·OEt2催化反应时,局部形成絮状物,最终无法固化;叔胺类催化剂TEA、HA对固化体系均有催化作用,但TEA由于位阻较大,催化效果并不理想,而HA具有特殊的笼状结构,位阻很小,催化效果较好。
HA催化GAP与TMPTGE的固化反应被认为是中间配合物机理反应[7],如图1所示。GAP中羟基与HA中三级胺形成氢键,进而与环氧基团形成配合物,这降低了开环加成反应的活化能,促进反应的进行。
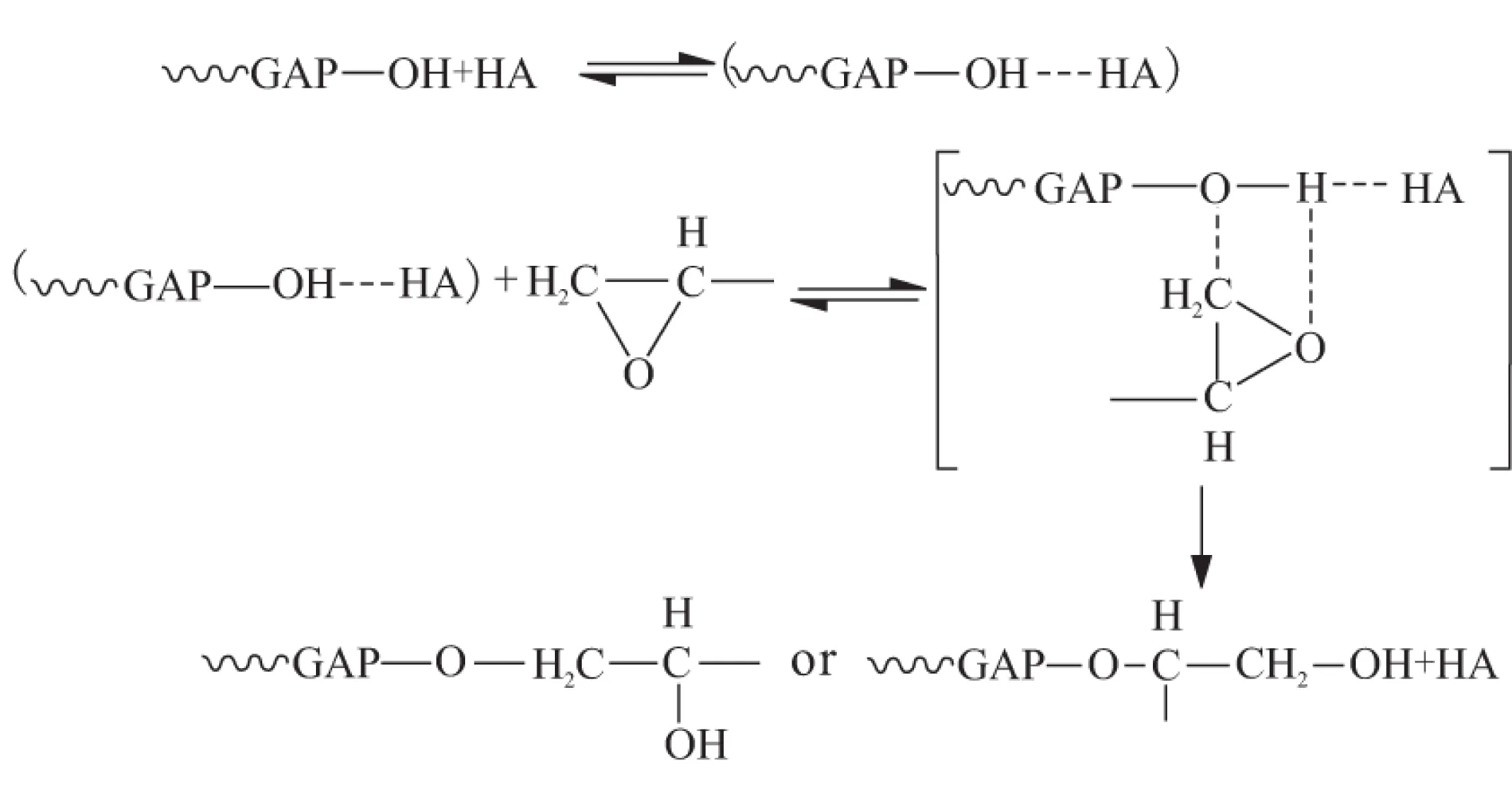
图1 HA催化GAP/TMPTGE的固化反应机理
在HA催化下,固化前后的GAP/TMPTGE体系样品通过FTIR进行表征,结果如图2所示。所表征固化体系R=3.0,HA含量为GAP质量的2%。
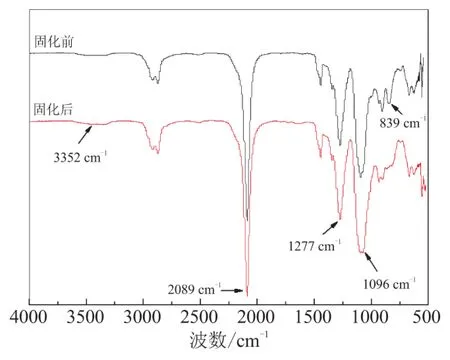
图2 GAP/TMPTGE固化前后的FTIR图
从图2可看出,固化前GAP/TMPTGE体系样品在3 352 cm-1附近有较弱的—OH伸缩振动峰,在2 089、1 277 cm-1处为—N3振动峰,在1 096 cm-1处为C—O伸缩振动峰,839 cm-1附近有明显的环氧基团特征吸收峰。样品固化后,2 089、1 277 cm-1处的—N3振动峰保持不变,而839 cm-1处的环氧基团特征吸收峰消失,1 096 cm-1处的C—O伸缩振动峰有所增强,表明体系中环氧基团发生开环加成反应,且反应较完全;由于羟基与环氧基开环加成时会生成另一个羟基,体系中羟基数目保持不变,因而3 352 cm-1附近的—OH特征吸收峰基本没有发生变化。
2.2GAP/TMPTGE胶片的力学性能
2.2.1固化参数R值对力学性能的影响
在HA的催化下,GAP的端羟基与环氧基团优先反应,但环氧化合物的自聚反应仍存在,而且GAP的端羟基与环氧基团反应生成的羟基也会进一步消耗环氧基团[7],因而体系R值选取应大于1。当催化剂HA含量为GAP质量2%时,固化时间为7 d,R值与其胶片的拉伸强度、延伸率之间的关系见图3。由图3可知,随固化参数R增大,胶片的断裂伸长率先增加、后降低,而拉伸强度却一直呈递增关系。R=2.0时,胶片的强度和延伸率均很差,当R=3.0时,断裂伸长率达到最大值98%,此时拉伸强度为0.67 MPa。由于体系中存在2种副反应,而且合成的弹性体为交联体系。因此,胶片的断裂伸长率并不理想。
为了探索R值改变所引起的固化体系网络结构改变对力学性能的影响,借助平衡溶胀法[8],测定了不同温度下固化产物的凝胶分数g,结果见表2。表2中还列出了不同R值对应的TMPTGE含量。
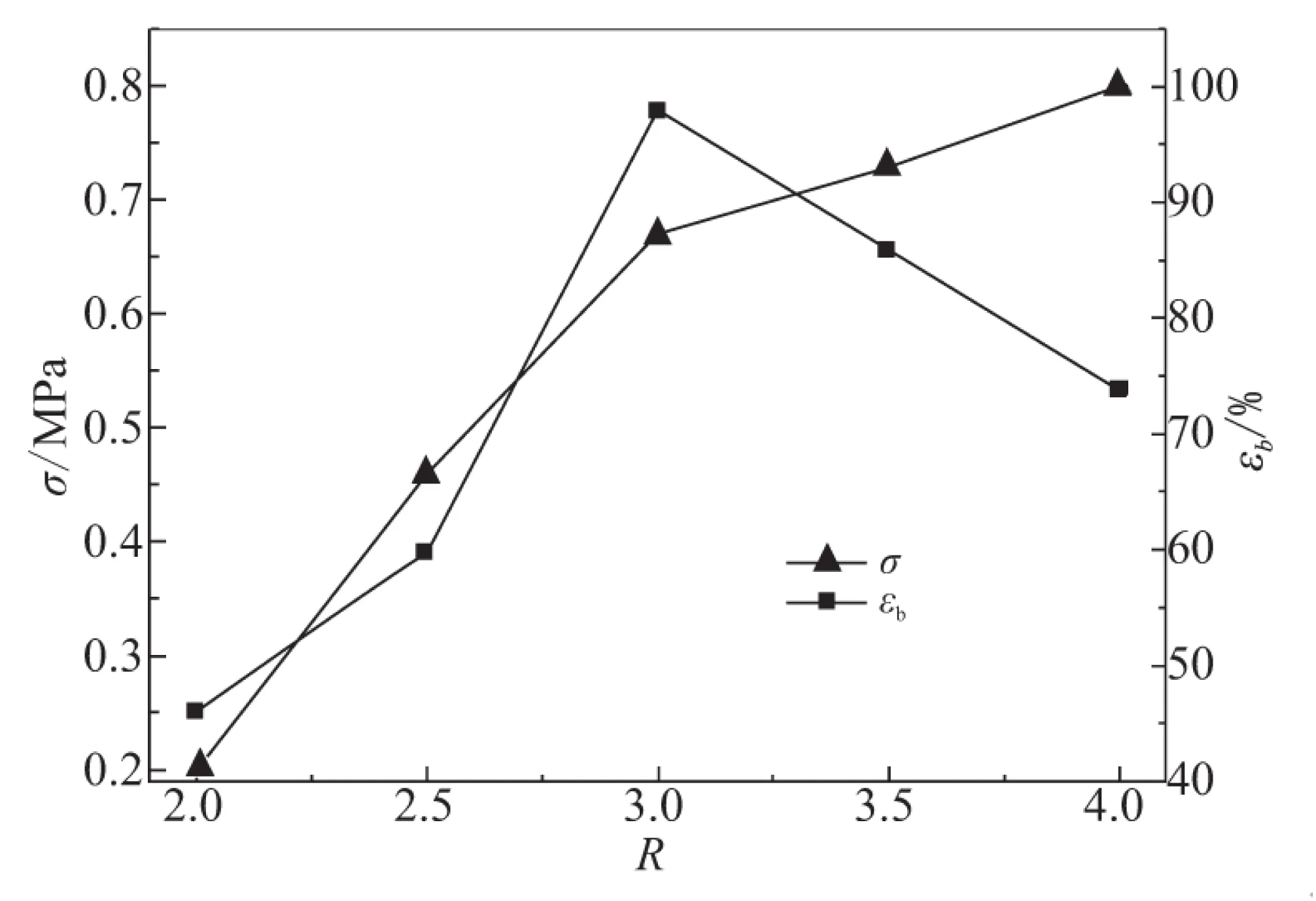
图3 R值对胶片力学性能的影响
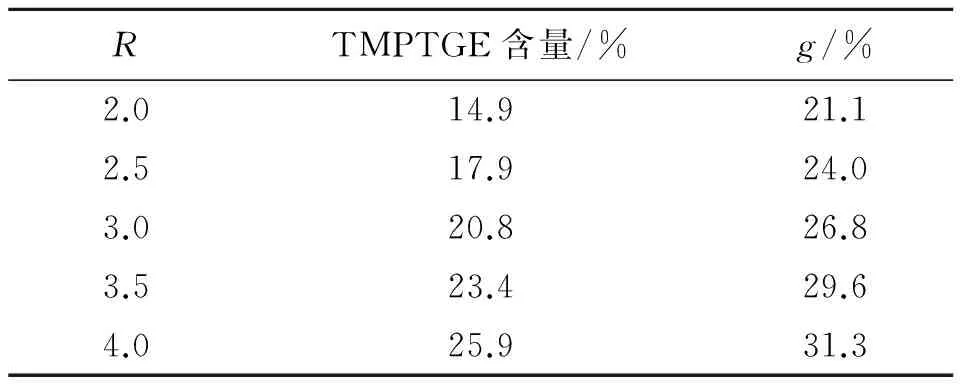
表2 不同R值胶片的凝胶分数
由表2可看出,随着固化参数R的增大,凝胶分数逐渐增加,固化体系中交联程度不断增加,因而胶片的拉伸强度不断增大。而交联程度的增大,也会导致断裂伸长率降低。因此,GAP/TMPTGE胶片的断裂伸长率会先增加、后降低。
另一方面,GAP/TMPTGE胶片的储能模量、损耗模量及玻璃化转变温度(Tg)等动态力学性能,可通过DMA实验进行表征,测试结果如图4所示。从图4中可知,GAP/TMPTGE胶片的储能模量与温度的关系曲线均只有一个转变过程,即损耗模量与温度的关系曲线上表现出来的单峰。通常定义损耗模量峰温为玻璃化转变温度,R值为2.0、3.0、4.0时,GAP/TMPTGE胶片的Tg分别为-33.3、-34.8、-34.8 ℃,而以4,4'-二苯基甲烷二异氰酸酯作为固化剂时,GAP胶片的玻璃化温度均高于-30.4 ℃[9]。环氧化合物作为固化剂时,不同R值胶片的玻璃化温度差异不大,但比异氰酸酯固化胶片的玻璃化温度更低,这可能是GAP/TMPTGE胶片不含氨酯基或脲基,没有所谓的“硬段”,链段基本不受固化剂影响的原因。此外,T

图4 GAP/TMPTGE胶片的DMA曲线
2.2.2固化时间对力学性能的影响
为研究固化时间对GAP/TMPTGE固化体系胶片力学性能的影响,选择固化参数R为2.0、3.0、4.0,分别制备多张胶片,在70 ℃下进行固化,每隔一定时间取出部分胶片测定其静态力学性能,结果如图5所示。

图5 固化时间对胶片力学性能的影响
从图5可看出,随着R值的增大,固化体系拉伸强度达到稳定时所需要的固化时间缩短。R=2.0时,胶片固化6 d后,拉伸强度基本保持在0.2 MPa左右。因此,固化温度为70 ℃时,延长固化时间为7 d,以保证各配方胶片的力学性能均达到稳定状态。
2.3GAP与TMPTGE固化反应动力学
表观活化能Ea是表征GAP与TMPTGE固化反应难易程度的参数,当固化体系获得的能量大于表观活化能时,固化反应才能顺利进行。因此,研究GAP/TMPTGE体系固化反应的表观活化能和反应级数等,对优化其固化工艺具有指导作用。
由前文可知,R=3.0时胶片的力学性能较好,选择此配方为测试样本。图6给出了不同升温速率下,该体系固化过程的DSC曲线。从图6可看出,随升温速率提高,放热速率增加,且放热峰的峰温向高温方向移动,峰形尖锐,说明固化反应速度随升温速率的升高而加快,固化时间缩短。
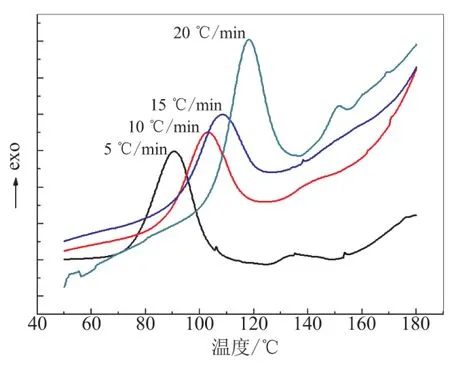
图6 不同升温速率DSC曲线
通过Kissinger方程和Crane方程,对不同升温速率下的DSC数据进行处理,可得固化体系的固化动力学参数[10-11]。体系固化反应的n级动力学模型及Kissinger方程、Crane方程分别对应于式(1)~式(3)。
(1)
(2)
(3)
式中α为固化度;A为指前因子;Ea为表观活化能,kJ/mol;R0为摩尔气体常数,R0=8.314 J/(mol·K); 为反应级数;β为升温速率,K/min;Tp为峰顶温度,K。
根据Kissinger方程和Crane方程,计算体系反应动力学参数所需的具体数据,如表3所示。根据表3中数据,以1/Tp为横坐标,以ln(β/T2)为纵坐标,利用Kissinger方程计算而得的ln(β/Tp2)与1/Tp的线性关系如图7所示。线性拟合可得直线斜率,即为-Ea/R0,截距为ln(AR0/Ea),进而求得表观活化能Ea=55.24 kJ/mol,指前因子A=3.705×105s-1。
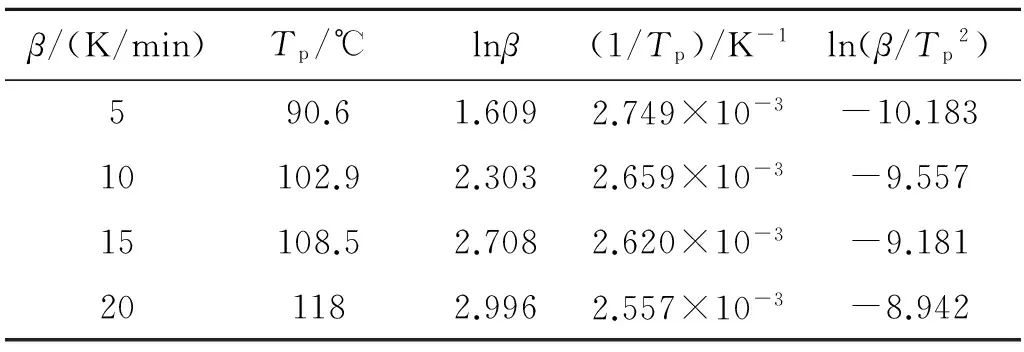
表3 DSC数据及初步拟合数据

图7 ln(β/Tp2)与1/Tp的关系
根据Crane方程,将lnβ与1/Tp线性拟合,可得直线斜率为-7 421.52。计算表2中不同升温速率下Tp的均值,并代入式(3)中2Tp项,该项约为斜率的10%,故不能省略,用2Tp对斜率进行校正,计算得反应级数n=1.00。
将所得Ea、A代入到固化动力学模型方程中,可得:
(4)
通过固化动力学的研究可看出,体系的固化程度取决于固化温度和固化时间。固化温度常用外推法求得,即升温速率为0时的特征固化温度。将表1中固化峰温线性拟合并外推,求得固化温度为83 ℃,但考虑到固化过程中的安全问题,选择70 ℃为固化温度。
2.4GAP/TMPTGE胶片的TG分析
热性能对于含能材料来说至关重要,因而采用热重法(TG)来研究GAP/TMPTGE体系固化成胶片后的热分解特性。图8为GAP及GAP/TMPTGE胶片的TG曲线。
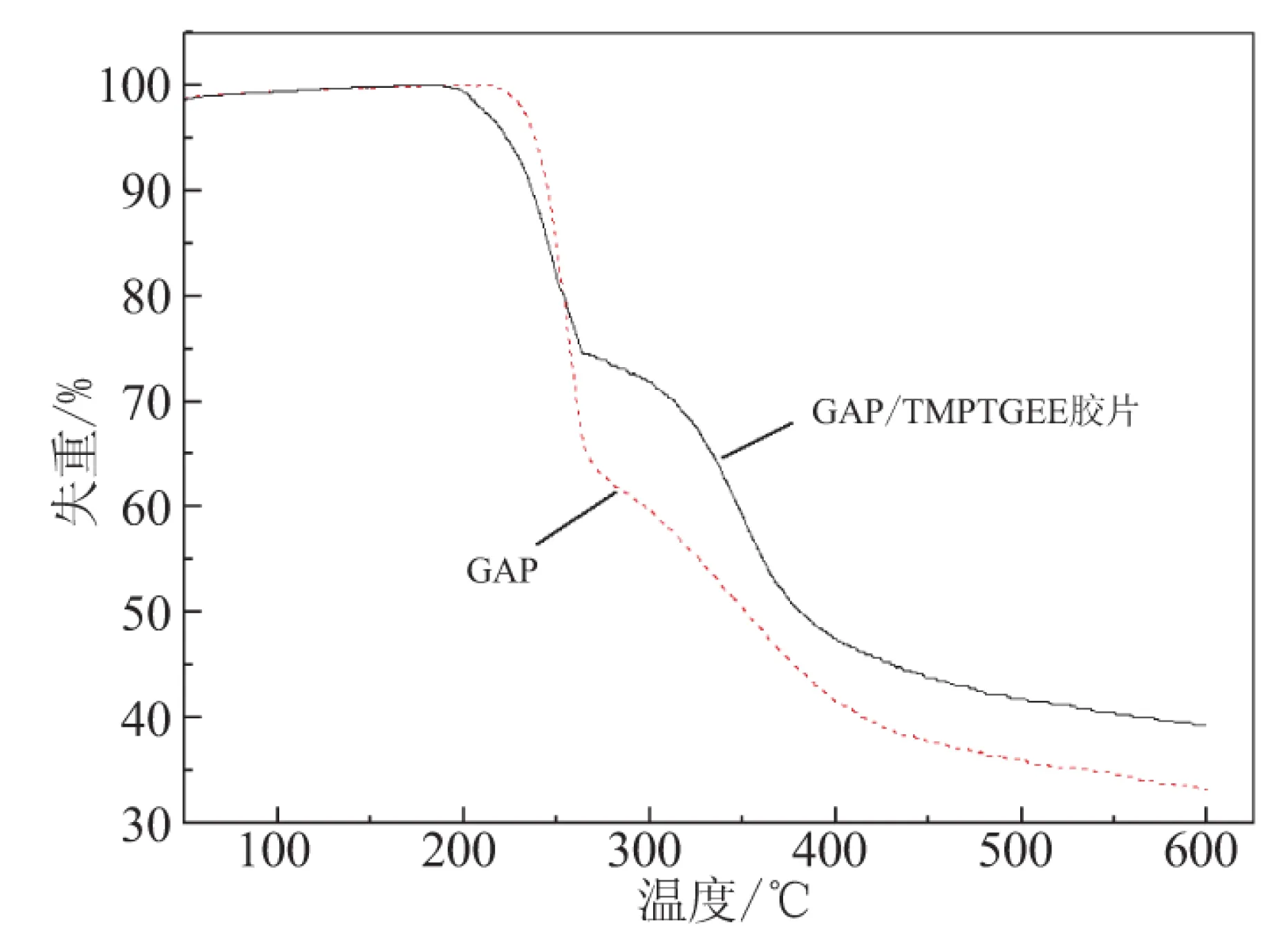
图8 GAP及GAP/TMPTGE胶片的TG曲线
由图8可知,GAP/TMPTGE胶片与GAP的热分解均分为2个阶段,具体热分解特性参数见表4。GAP/TMPTGE胶片热分解的第1阶段可认为和GAP一样,为—N3分解,但分解峰温略有提前,质量损失明显小于GAP;第2阶段为骨架分解,分解峰温推后,质量损失大于GAP;2个过程整体质量损失明显小于GAP。GAP/TMPTGE胶片分解温度没有显著提前,说明TMPTGE作为固化剂时,体系的热稳定性与GAP相近,而整体质量损失较小,则应是TMPTGE的加入使GAP中含能基团比例减小所致。
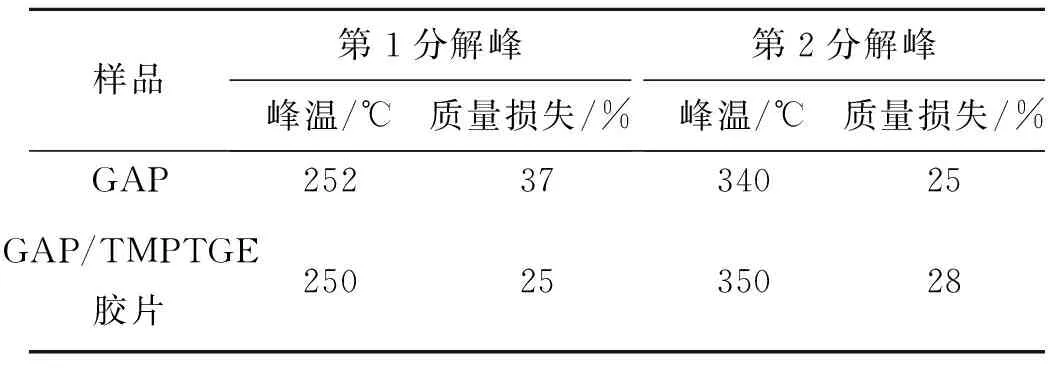
表4 GAP及GAP/TMPTGE胶片热分解特性参数
3 结论
(1)HA催化GAP/TMPTGE固化的效果较好,在其催化下,GAP/TMPTGE在70 ℃下固化得到交联型弹性体材料,固化时间为7 d,可保证固化充分。
(2)随着固化参数R的增大,胶片的断裂伸长率先增加、后降低,而拉伸强度却一直呈递增趋势。R=3.0时,GAP/TMPTGE胶片的断裂伸长率达到最大值98%,此时拉伸强度为0.67 MPa,玻璃化转变温度为-34.8 ℃。采用非等温DSC法进行固化动力学研究表明,反应的表观活化能Ea=55.24 kJ/mol,指前因子A=3.705×105s-1,动力学机理函数f(α)=(1-α)n,其中n=1.0。
(3)GAP/TMPTGE胶片热分解均分为2个阶段,与GAP热分解情况相近,第1阶段分解峰温为250 ℃,质量损失25%;第2阶段分解峰温为350 ℃,质量损失28%。
(4)环氧化合物作为GAP固化剂时,固化体系对水分不敏感,易吸湿的氧化剂AN、ADN等可尝试用于该体系,用以进一步提高推进剂的性能。但该体系力学性能不理想,寻找选择性更高、催化效率更好的固化催化剂及更加合适的固化条件是进一步改进的方向。
[1]侯林法.复合固体推进剂[M].北京: 中国宇航出版社,1994: 32-33.
[2]Figovsky O L,Shapovalov L D.Features of reactionamino-cyclocarboante for production of new type non-isocyanate polyurethane coatings[J].Macromolecular Symposia,2002,187: 325-332.
[3]张晗昱,翟进贤,杨荣杰.端叠氮基聚醚的合成及与多炔固化剂的交联反应[J].火炸药学报,2012,35(5): 45-48.
[4]Frankel M,Grant L,Flanagan J.Historical development of GAP[R].AIAA 80-2307.
[5]蔚红建,付小龙,樊学忠,等.GAP及GAP推进剂研究新进展[J].飞航导弹,2010(11): 90-93.
[6] Keicher T,Kuglstatter W,Eisele S,et al.Isocyanate free curing of glycidyl-azide-polymer (GAP) with bis-propargyl-succinate[C]//39th Int.Annu.Conf.of ICT,2008.
[7]陈平.环氧树脂及其应用[M].北京: 化学工业出版社,2004.
[8]宋永莱,聂海英,马新刚.NEPE推进剂凝胶分数测定方法研究[J].固体火箭技术,2002,25(4): 35-37.
[9]左海丽.GAP基含能热塑性弹性体研究[D].南京: 南京理工大学,2011.
[10]Kissinger H E.Reaction kinetics in differential thermal analysis[J].Analytical Biochemistry,1957,29(11): 1702-1706.
[11]Crane L W,Dynes P J ,Kaelble D H.Analysis of curing kinetics in polymer composites[J].Polym.Lett.Ed.,1973,11(8): 533.
(编辑:刘红利)
Preparation and properties of GAP/epoxide curing system
ZHENG Qi-long,ZHOU Wei-liang,XIAO Le-qin,JIAN Xiao-xia
(School of Chemical Engineering,Nanjing University of Science & Technology,Nanjing210094,China)
Glycidyl azide polymer(GAP) was cured by trimethylolpropane triglycidyl ether(TMPTGE) in the presence of a tertiary amine catalyst(HA). The effects ofRand cure time on the mechanical properties of the GAP/TMPTGE film were characterized by tensile test and DMA. The kinetic characteristics of this curing reaction were studied thoroughly by means of DSC, and thermal properties were studied by TG. The results indicate that the breaking elongation of the film increases at first then decreases and the tensile strength increases straightly with the increase of theR. WhenRis 3.0, the breaking elongation of the film is 98%, the tensile strength is 0.67 MPa and the glasstransition temperature of the film is -34.8 ℃. The TG experimental results show that the decomposition of cured film can be divided into two steps, the decomposition peak temperatures of which are 250 ℃ and 350 ℃ respectively.
glycidyl azide polymer(GAP);epoxide;mechanical properties;thermal properties
2015-03-18;
2015-04-27。
郑启龙(1989—),男,博士生,主要从事固体推进剂研究。E-mail:zhenglong2577@163.com
周伟良(1963—),男,研究员,主要从事含能材料相关研究。E-mail:wlzhou331@163.com
V512
A
1006-2793(2016)04-0497-06
10.7673/j.issn.1006-2793.2016.04.009
猜你喜欢
大电机技术(2022年5期)2022-11-17
含能材料(2022年8期)2022-08-13
军民两用技术与产品(2022年7期)2022-08-06
兵器装备工程学报(2020年11期)2020-12-16
石材(2020年8期)2020-10-28
火炸药学报(2019年2期)2019-05-05
筑路机械与施工机械化(2017年6期)2017-07-10
大陆桥视野·下(2016年9期)2017-05-08
科技创新与应用(2017年7期)2017-03-27
航天制造技术(2016年6期)2016-05-09
