
连接基团对双核二茂铁分子结构参数影响的计算研究
2016-11-03 02:39:46姜本正
固体火箭技术 2016年2期
姜本正,张 炜,杨 军,邓 蕾,俞 艳,鲍 桐
(1.国防科技大学 航天科学与工程学院,长沙 410073;2.中科院上海有机化学研究所,上海 200000)
连接基团对双核二茂铁分子结构参数影响的计算研究
姜本正1,张炜1,杨军2,邓蕾1,俞艳2,鲍桐1
(1.国防科技大学 航天科学与工程学院,长沙410073;2.中科院上海有机化学研究所,上海200000)
采用Gaussian商用软件密度泛函理论(DFT)中的B3LYP方法,在6-311G*基组水平上,对双核二茂铁进行几何优化,获得平衡构象。在此基础上,计算了二茂铁间连接基团对双核二茂铁分子偶极距、前线轨道能隙、原子电荷及键长等分子参数的影响。结果表明,连接基团越复杂,双核二茂铁分子前线轨道能隙减小,分子整体稳定性下降,弱键位于连接基团内;本研究范围内,最为稳定的双核二茂铁为二二茂铁基甲烷。
密度泛函理论;二茂铁衍生物;连接基团;双核
0 引言
自1951年首次报道以来[1],二茂铁及其衍生物因其独特的性能引起了化学界、材料学界的普遍关注[2-4]。二茂铁衍生物具有较高的铁含量,而铁元素被认为是AP/Al/HTPB推进剂的理想催化成分,因而二茂铁衍生物作为燃速催化剂广泛用于AP/Al/HTPB推进剂中。与单核二茂铁衍生物相比,双核二茂铁衍生物具有更高的铁含量、抗迁移优良等特点,是该类燃速催化剂的发展方向。目前,应用最广泛双核二茂铁衍生物燃速催化剂是Catocene(国内类似结构称GFP),它具有铁含量高、燃速催化效率高、不易迁移等优点[5-7]。但其与超细AP相互作用时,机械感度过高,严重影响该类高燃速推进剂的生产、运输及使用[8-9]。因此,设计和合成新型钝感、高效的双核二茂铁燃速催化剂,成为近些年的研究热点。
本文以双核二茂铁为主要研究对象,采用密度泛函理论,考查二茂铁间连接基团对双核二茂铁偶极距、前线轨道能隙、原子电荷及键长等分子参数的影响,以预测分子的稳定性,为设计新型钝感双核二茂铁衍生物燃速催化剂提供理论依据。
1 理论方法
计算分子结构的理论方法主要有Hatree-Fork (HF)方法[10-12]和密度泛函理论(DFT)等[13-16]。大量研究表明[17-18],DFT方法被广泛用于计算含过渡金属原子的分子构型,并在精度上高于HF方法。因此,本文基于Gaussian[19]商用软件,采用密度泛函理论(DFT)中B3LYP方法[20]的6-311G*基组,计算二茂铁分子及其衍生物的平衡构象及分子结构特性参数,计算由PC机完成。二茂铁分子中各类键长与文献报道实验值的比较结果如表1所示。
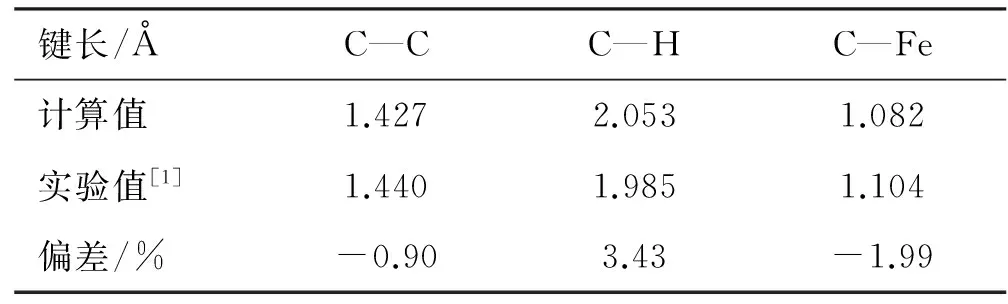
表1 二茂铁分子理论计算结果和实验值
由表1可知,采用密度泛函理论(DFT)中B3LYP方法的6-311G*基组,计算得到的结果与实验值十分接近,最大偏差仅为3.43%,说明此方法适用于二茂铁体系的理论研究。
本文称连接2个二茂铁分子之间的基团为连接基团。在此基础上,采用上述计算方法,以二茂铁为基准分子,重点研究2个二茂铁间连接碳的基团对双核二茂铁分子平衡构象及分子结构参数(偶极距、前线轨道能隙、原子电荷及键长等)的影响。
Fe元素含量是衡量二茂铁衍生物类燃速催化效率的重要指标之一。因此,连接基团不能过于复杂。除较高的Fe含量外,用于AP/Al/HTPB固体推进剂中的二茂铁衍生物还需要考虑催化剂分子与推进剂固化体系的相容性。因此,一般在设计双核二茂铁衍生物时,不考虑O、S等杂原子及具有强反应活性的—OH和—COOH等基团。基于上述考虑,本研究选取的双核二茂铁衍生物如表2所示,其特点是R1为亚甲基,为双核二茂铁的最简单结构;R2和R3的连接碳原子分别有1个和2个甲基。表2中,Fc为二茂铁基团。
2 结果与讨论
2.1单核二茂铁与双核二茂铁的比较
计算得到的二茂铁分子及R1分子平衡构象如图1所示。

表2 二茂铁衍生物分子分子结构

(a)二茂铁
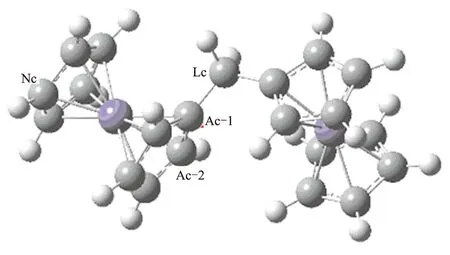
(b)R1
双核二茂铁分子中原子数众多,尤其是C、H原子。为对分子中各原子进行区分,将其进行编号。编号原则为:
(1)2个二茂铁间连接C原子为LC,连接碳原子上所接甲基C原子为LCplus-n,n为序号。
(2)与连接基团相连的二茂铁戊环(一侧为A,另一侧为B)中C原子为AC-n(或BC-n)。其中,n为序号。当戊环C原子直接与LC相连时,n=1;其余C原子序号按顺时针编排。
(3)计算结果表明,未与连接基团相连的2个戊环中各原子受连接基团的影响较小。因此,该2个戊环中C原子不作区分,统称NC。
(4)H原子编号取与其连接的C原子编号。
(5)Fe原子不编号。
(6)二茂铁分子对称性好,不对其进行区分。
计算得到的重要分子结构参数如表3所示。
从图1和表3可看出:
(1)二茂铁分子结构对称,偶极矩为0。说明正负电荷中心重合,分子稳定性好。
前线轨道能隙是衡量分子整体稳定性的最直接表征参数。能隙越大,分子越稳定;反之亦然。二茂铁分子的前线轨道能隙为0.197a.u.,与固体推进剂常用粘合剂聚丁二烯体系的重复单元*n*(0.185a.u.,同条件计算值)相比,更稳定。

表3 二茂铁与R1双核二茂铁分子的分子结构参数
键长可表征同类型化学键的强弱,可发现分子弱键的位置。同类型化学键键长越短,键能越高,化学键越稳定;反之,化学键越薄弱,即该键易断裂。计算结果表明,二茂铁分子戊环中的C—C键长均相等,且介于孤立单键(1.514 Å)和双键(1.326 Å)之间,表明戊环具有类似苯环(C—C键长1.395 Å)的共轭特征;但由于五元环张力大于六元环,因此二茂铁分子中的茂环稳定性低于苯环。
(2)尽管在2个二茂铁之间连接了一个亚甲基,但R1分子失去了分子的对称性,偶极距为0.3661 D;其分子的前线轨道能隙下降至0.191 a.u.,说明其分子的稳定性比二茂铁差;与二茂铁分子相比,与连接亚甲基相连的2个戊环中C—C键长较均有一定增加,最大键长为AC-1—AC-2和AC-1—AC-5,说明戊环的稳定性变差是受取代基的影响;且距离取代基越近,影响越明显;但尽管如此,这2个键的键长仍比孤立的C—C单键键长短,但明显长于原戊环内C—C键,说明在这个双核二茂铁分子中,A(B)C-1—LC属于弱键,但其稳定性高于孤立C—C键,说明R1分子仍具有一定的稳定性。
二茂铁分子及R1分子的电子云分布如图2所示。
从图2可看出,二茂铁分子电荷分布较为均匀。正电中心位于分子中Fe原子处;负电中心有2个,分别位于戊环的几何中心。R1号分子负电核中心依旧主要分布于戊环中心位置,正负电荷中心数目增多。对各原子所带电荷数进行计算,结果见表4。

(a)二茂铁

(b)R1

二茂铁原子种类原子电荷/e原子种类R1原子电荷/eC-0.158LC-0.258AC-0.012,-0.155~-0.176BC-0.005,-0.155~-0.176NC-0.154~-0.159H0.100H0.097~0.101Fe0.580Fe0.590,0.573
从表4可看出,二茂铁分子中同种原子的电荷相同,Fe和H原子均带正电,C原子带负电。因此,带正电基团攻击时,一般攻击C原子;带负电基团一般攻击H原子。但由于C、H所带电核较少,反应活性极低;尽管Fe原子具有较高的正电荷,但由于双戊环的位阻效应,在分子解构前很难进行反应;而分子解构需要较高的能量,因此二茂铁分子相对性质稳定。
因连接基团的影响,R1分子中同种原子的电荷出现了差异。其中,连接C原子连接了2个电负性较弱的H原子及2个富电子的戊环,因此带有数目最多的负电荷;且由于自身空间位置较为裸露,易为带正电核基团攻击而发生反应。对于与连接C原子相连的戊环中的2个C原子AC-1和BC-1,由于自身部分电荷转移至连接基团LC原子,自身所带负电荷数下降;同时,连接戊环中C原子的电荷出现较大的波动。即除AC-1和BC-1外,C原子的电荷在-0.155~-0.176 e范围变化。非连接戊环C原子受连接基团影响较小,表现在NC原子电荷波动范围不大,与二茂铁分子电荷相近。H原子电荷也无较大变化。2个Fe原子因所属二茂铁基团的空间位置不同,其受连接基团的影响强弱也不同。
综上所述,与二茂铁分子相比,双核二茂铁分子的稳定性出现一定程度的下降;弱键位于连接C原子与戊环中C原子之间的化学键即LC—AC-1(BC-1);连接基团中心C原子是一个潜在的反应活性中心。
2.2连接基团对双核二茂铁衍生物分子结构参数的影响
计算得到R2和R3分子的平衡构象见图3。计算得到2个双核二茂铁的重要分子结构参数见表5。

(a)R2

(b)R3

编号连接基团偶极矩/DΔELUMO-HOMO/a.u.键长/ÅAC—AC'A(B)C-1—LCLC—LCplusR1—CH2—0.36610.1911.428~1.4321.504,1.505—R2—CH(CH3)—0.33410.1911.426~1.4331.515,1.5201.544R3—C(CH3)2—0.35680.1891.427~1.4351.527,1.5331.541,1.551
从图3和表5可看出,随连接C原子上甲基数目增加,偶极矩出现不规则变化;前线轨道能隙出现下降趋势,说明引入的—CH3增强了分子的空间位阻作用,分子反应活性升高,造成分子整体稳定性下降。
随连接C原子上甲基数目的增加,戊环内C—C键的键长出现上升的趋势,说明由于连接基团上—CH3数目的增多,分子内的空间位阻增大,导致戊环发生畸变的程度加大,稳定性下降;LC—AC-1(BC-1)键长大幅上升,说明连接基团与戊环的键合减弱,反应活性增强;连接基团中,LC—LCplus键长大于孤立单键(1.514 Å),说明其结合更弱,即与LC—AC-1(BC-1)相比,反应活性更强,是新的反应活性中心;另外,在R3分子中,两LC—LCplus键长出现差异。这是由于其与二茂铁基团的相对空间位置不同所造成。
连接基团不同的双核二茂铁分子中原子电荷如表6所示。从表6可看出,随取代基—CH3的引入,双核二茂铁分子中C原子电荷发生了规律性的变化:连接C原子所带负电荷数下降,与其相连的—CH3C原子具有较高的负电荷,成为新的负电荷集中点,易于发生反应。且随—CH3基团数目增多,与连接C原子相连的—CH3中碳原子的最高负电荷量上升,说明其作为活性中心的反应性增强;与连接C原子相连的戊环中AC-1(BC-1)原子负电荷量出现下降趋势,甚至出现带正电荷的情况,说明其周围的电子云密度下降,戊环整体的大π共轭效应减弱,戊环稳定性下降。与连接C原子相接的戊环AC-1或BC-1原子电荷的不平衡性与其所处空间位置有关。H原子的原子电荷差距较小,一般戊环上H的电荷略高于连接基团烷基的H原子,这是由于戊环的大π共轭效应导致。随—CH3数增加,Fe原子电荷的正电荷数值减小。
由原子电荷的变化可看出,当连接C原子上引入—CH3后,活性中心发生变化,即由原来的连接C原子转移至与—CH3中的C原子;且在R3分子中,由于空间位置不同,2个LCplus原子也有一定的差异。从稳定性角度考虑,R1分子优于R2和R3分子。
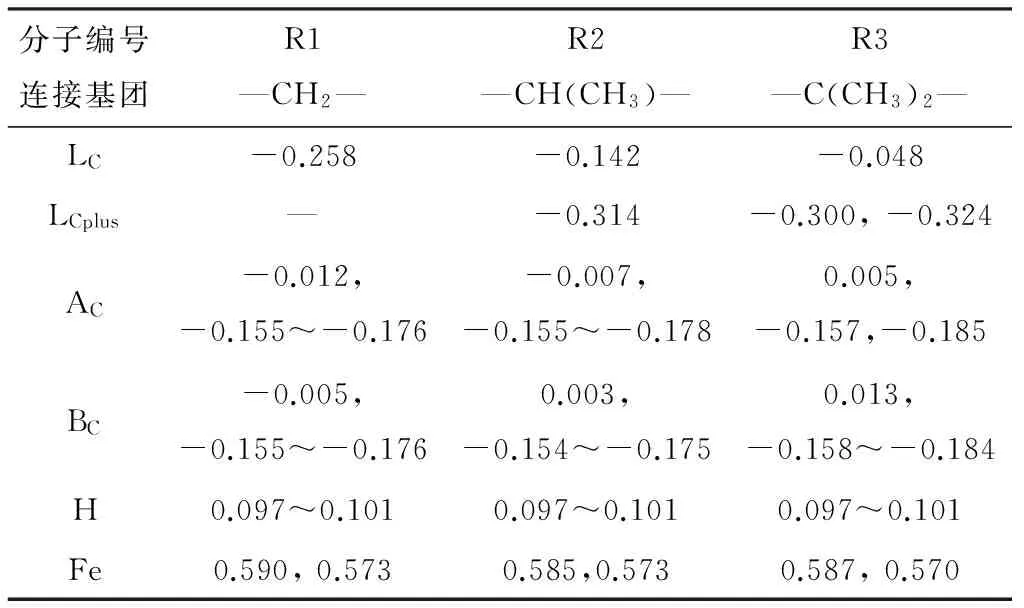
表6 连接基团对双核二茂铁分子原子电荷的影响
3 结论
(1)采用密度泛函理论(DFT)中B3LYP方法中的6-311G*基组,对双核二茂铁分子的计算具有可信性,可对分子性质进行预测。
(2)连接基团越复杂,前线轨道能隙减小,双核二茂铁分子的稳定性下降,弱键出现于与连接C原子相接的化学键。
(3)本研究范围内,最为稳定的双核二茂铁为R1,即二二茂铁基甲烷。
[1]Kealy T J,Pauson P L.A new type of organo-iron compound[J].Nature,1951,4285:1039-1040.
[2]Cowan D O,Park J,et al.Semiconducting polymers.mixed valence ferrocene-ferricenium polymers[J].Am.Chem.Soc.,1971,94(14):5110-5112.
[3]Kertesz M.Handbook of organic conductive molecules and polymers[M].New York:John Wiley & Sons Ltd.,1997.
[4]Hmynen M,Yassar A,Percheron-Guegan A,et al.Magnetic properties of ferrocene-based conjugated polymers[J].Adv.Mater.,1994,7(8):564-568.
[5]段军鸿,崔瑞禧,杨军.新型抗迁移二茂铁燃速催化剂的分析与试验[C]//中国宇航学会固体推进第29届学术年会论文集.宁波,2012.
[6]唐劲松,丁宏勋.丁羟推进剂的高效燃速催化剂[J].化学推进剂与高分子材料,2004,201:8-11.
[7]杨军,邓敏智.新型不迁移的二茂铁燃速催化剂的研制[J].固体火箭技术,2007,30(1):44-56.
[8]王宁,赵孝彬,罗岚,等.AP/二茂铁衍生物混合体系的热感度研究[J].固体火箭技术,2007,34(2):207-210.
[9]张炜,杨军,俞艳,等.二茂铁种类对超细AP/二茂铁体系感度的影响[J].含能材料,2011,19(6):627-631.
[10]Hartree D R.Wave mechanics of an atom with a non-coulomb central field.I.Theory and methods.Ⅱ.Some results and discussion.Ⅲ.Term values and intensities in series in optical spectra[J].Prog.Camb.Phil.Soc.,1928,24:89-110.
[11]Fock V Z.The initial degrees of freedoms of the electron[J].J.Phys.,1931,68:522-534.
[12]Roothan C C J.New developments in molecular orbital theory[J].Rev.Mod.Phys.,1951,23:69-89.
[13]Thomas L H.The calculation of atomic fields[J].Proc.Camb.Phil.Soc.,1927,23:542-548.
[14]Fermi E.Statistical calculation of the Rydberg correction of the system[J].J.Phys.,1928,49:550-554.
[15]Slater J C.A simplification of the Hartree~Fock method[J].J.Phys.Rev.,1951,81:385-390.
[16]Hohenberg P,Kohn W.Inhomogenous electron gas[J].J.Phys.Rev.,1964,B136:864-871.
[17]刘艳红,赵景祥,刘继红,等.二茂铁取代物的理论研究[J].哈尔滨师范大学自然科学学报,2006,22(3):67-69.
[18]邓嘉莉,宁顺群,廖显威.二茂铁的结构探讨与核磁共振碳谱计算[J].西南民族大学学报(自然科学版),2005,31(2):115-117.
[19]Frisch M J,Trucks G W,Schlegel H B,Scuseria G E,Robb M A,Cheeseman J R,Montgomery J A Jr,Vreven T,Kudin K N,Burant J C,Raghavachari K,Foresman J B,Ortiz J V,Cui Q,Baboul A G,Clifford S,Cioslowski J,Stefanov B B,Liu G,Liashenko A,Piskorz P,Komaromi I,Martin R L,Fox D J,Keith T,Al-Laham M A,Peng C Y,Nanayakkara A,Challacombe M,Gill P M W,Johnson B,Chen W,Wong M W,Gonzalez C,Pople J A(2003) Gaussian 03.Gaussian,Inc.,Pittsburgh.
[20]Becke A D.Density functional thermochemistry.III.The role of exact exchange[J].J.Chem.Phys.,1993,98:5648-5652.
(编辑:刘红利)
A theoretcal study on effect of link-group on double-core ferrocene derivatives
JIANG Ben-zheng1,ZHANG Wei1,YANG Jun2,DENG Lei1,YU Yan2,BAO Tong1
(1.School of Aerospace Science and Engineering,National University of Defense Technology,Changsha410073,China; 2.Shanghai Institute of Organic Chemisty,Chinese Acedamy of Science,Shanghai200000,China)
Based on Gaussian communication software,the effect of link-group on double-core ferrocene was studied by DFT theory and B3LYP method.The results show that B3LYP method is suitable for investigation in this study.Link-group greatly changes the structure of double-core ferrocene.With the number of group —CH3increasing,the energy gaps of frontier molecular orbital and the stability of the molecular are decreased.Weak point of the molecular is located in the link-group.The double ferrocenyl-methane is the most stable molecular in this study.
DFT;ferrocene derivatives;substituent;double-core
2015-01-15。
国家自然科学基金委员会和中国工程物理研究院联合基金(NSAF)(U1230102)。
姜本正(1986—),男,博士生,研究方向为固体推进剂。E-mail:benzhengforever@126.com
V512
A
1006-2793(2016)02-0220-05
10.7673/j.issn.1006-2793.2016.02.012
猜你喜欢
物理学报(2021年12期)2021-07-01 09:42:56
数学物理学报(2020年6期)2021-01-14 01:00:46
火炸药学报(2019年2期)2019-05-05 08:33:56
固体火箭技术(2016年5期)2016-11-03 05:35:29
武汉工程大学学报(2016年1期)2016-04-07 02:01:11
中国塑料(2015年5期)2015-10-14 00:59:47
海军航空大学学报(2015年4期)2015-02-27 13:45:52
火炸药学报(2014年5期)2014-03-20 13:17:53
火炸药学报(2014年1期)2014-03-20 13:17:28
无机化学学报(2014年9期)2014-02-28 17:33:11
