
对结构化学中价键理论教学的若干思考
2016-11-01 06:44:08侯华黄智超王宝山
大学化学 2016年9期
侯华 黄智超 王宝山
(武汉大学化学与分子科学学院,武汉 430072)
对结构化学中价键理论教学的若干思考
侯华黄智超王宝山*
(武汉大学化学与分子科学学院,武汉430072)
通过追踪原始文献,分析结构化学中价键理论部分的教学内容,特别是对氧气分子基态电子结构的处理,发现价键理论不仅能够正确解释或预测分子结构,而且对芳香性、铁磁性等化学键本质的描述表现更优。
价键理论;化学键;氧气;芳香性
1 引言
价键(Valence Bond,VB)理论与分子轨道(Molecular Orbital,MO)理论是预测分子结构与化学键本质的代表性模型,也是结构化学课程教学的重要内容[1-10]。两种理论的区别在于构造波函数的方式不同。早在1916年,Lewis[11]就提出了“电子对键”的思想。1927年,Heitler与London[12]在处理氢分子结构时首次采用两个氢原子基态电子波函数的乘积表示电子对键,通过共振结构波函数的线性组合获得薛定谔方程的解。随后经过Sugiura、Rosen、Hylleraas、特别是我国量子化学先驱之一王守竞等[13]的发展,Pauling[14]在1931年建立了较为完善的电子对键与杂化轨道理论模型,随后以电子配对形成定域化学键为核心思想的价键理论,凭借其既直观又能定量计算的优势,得以在化学领域迅速推广应用。与此同时,Mulliken[15]提出了分子轨道理论,虽然同样受Heitler与London处理氢分子的启发,但是他们将分子中的每一个电子分开来考虑,即所谓的单电子波函数近似,把分子的多电子Schrodinger方程转化为多个单电子运动方程。由于数值计算的困难,直到六十年代,分子轨道理论才随着计算机技术的飞速发展和计算软件的普及得以广泛应用。特别是近年来大量几乎“黑盒子”式的商业模拟计算软件的涌现,使得分子轨道理论逐渐取得了在量子化学领域的统治地位。与此同时,部分面向本科生的结构化学教材也存在着一些关于价键理论缺点的说法[5-8]。例如,价键理论无法正确得到简单氧气分子基态的结构与性质,特别是实验测定的顺磁性;对于环丁烯、C5H5+的结构、甲烷分子的光电子能谱等问题价键理论也无能为力。相反地,这些价键理论的问题,分子轨道理论则可以给出正确的预测或解释。诸如此类看似矛盾的论述不仅是对结构化学学科严谨性与科学性的损害,而且不利于结构化学的教学工作,更让刚开始学习结构化学的学生感到困惑,严重影响了他们学习结构化学、从理论角度思考化学问题的兴趣。因此,我们认为很有必要对价键理论进行深入探讨,澄清事实真相,恢复对价键理论应有的认识与评价。
本文以氧气分子为例,结合文献调查与分析,对其基态结构与化学键特征进行了详细讨论。证明价键理论不仅能够正确定性描述氧气分子的成键与顺磁特性,而且还可以定量计算得到其化学键本质。然后通过对环丁烯芳香性判断以及锂金属团簇的不成对铁磁性键两个具体例子,表明价键理论在处理复杂分子体系结构与性质方面同样优越。最后通过分析价键理论与分子轨道理论的本质,探讨了两种理论的一致性。
2 价键理论对基态氧气分子结构的解释
光谱实验证明基态氧气(O2)分子为三重自旋态,即包含两个未成对电子,呈顺磁特征。根据分子轨道理论,两个O原子的2p轨道可以形成两个π成键轨道与两个π*反键轨道。按照Aufbau原理、Pauli不相容原理以及Hund规则,形成O2分子时有两个电子分别填充在两个π*反键轨道上且自旋平行,故很自然地得到O2基态为三重态的结论[16]。若采用价键理论,根据电子配对方法,由O原子的电子组态出发,将其配对形成完美的O=O双键结构,分子中不存在单电子,从而得到O2分子基态为单重态的结论。
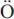

图1 氧原子(A)与氧气分子的结构(B,C)
我们发现可以根据直观的电子配对成键图像理解O2分子的结构。根据Pauling的价键理论和s-p杂化近似波函数的构成条件,每个O原子有6个价电子(2s22p4),当组合形成O2分子时,除用于形成两个σ键与一个σ*反键外,共剩余6个电子,将填充于两个相互垂直的π平面(px-px,py-py)内,则共存在4种可能的填充方式,如图2所示。

图2 氧原子的6个p电子在px-px、py-pyπ平面上的4种填充方式
从图2可以很明显看出:A与B两种填充方式得到两个π平面上各含3个电子的双自由基(Biradical),而C与D两种填充方式则得到闭壳层单重态结构。通过简单的键级分析可知,A/B填充方式的键级均为0.5+0.5=1,而C/D填充方式的键级亦为1+0或0+1=1,说明O2存在4种互变异构的共振结构。另一方面,通过对C/D的进一步观察,不难发现键级为0的py-py平面上存在2对孤对电子的Pauli排斥作用,表现为反键特征,而A/B结构存在较强的电子离域特征。因此,C/D的能量应该比A/B更高,说明O2分子的基态应以A/B两种填充方式占主导地位。A/B中2个单电子可以形成单重态或三重态波函数,由于单电子轨道的正交性质与交换相关作用,三重态能量更低,所以氧气分子呈双自由基结构。如果将图2所示的4种共振结构构造成为价键波函数,即可进行定量计算,获得O2分子的价键结构与能量。1973年Goddard等[20]曾使用广义价键理论(Generalized Valence Bond,GVB)方法得到了正确的O2基态。1991年McWeeny[21]采用价键理论计算了O2的基态结构,其价键波函数包含两种离子共振结构,发现价键理论要比基于自洽场的分子轨道理论更为精确,也更能直观地揭示O2分子双键的本质。
从以上讨论可以看出,无论是Lewis结构还是Pauling的价键结构,无论定性分析还是定量计算,都能够对O2分子的基态电子结构进行解释,文献中并不存在价键理论无法正确描述O2化学键的依据。
3 价键理论与芳香性及不成对铁磁性键
芳香性是一个重要的化学概念,而分子轨道理论在分子芳香性预测方面非常成功。结构化学中用于判断单环共轭多烯芳香性的4n+2或4n规则即建立在Huckel分子轨道理论基础之上[22],判断多环共轭多烯的芳香性与反芳香性则常用Craig的分子轨道模型[23]。然而,这些基于分子轨道理论的判断方法也会出现误判问题。根据Craig规则,环丁二烯不具有芳香性,与实验观测相符;联环丁二烯包含通过两个四元环中心的对称轴,Craig规则预测它们是芳香性化合物,但是实验结果证实联环丁二烯为反芳香性,分子轨道理论无法正确解释这一观测结果,即使采用高级电子相关计算也无能为力。1998年Zilberg与Hass[24]基于价键理论提出了双态模型,能够很好地预测C2nH2n(n≥2)共轭烯烃的芳香性。对于环丁二烯分子的价键理论处理如图3所示。

图3 环丁二烯的双态共振结构与能级示意图
可以看出,L与R两种共振结构是等价的,可以用二者的线性组合构造价键波函数,其中L+R称为“in-phase”波函数,L-R称为“out-of-phase”波函数。根据价键理论的计算结果,可知:当有偶数个电子对时,L-R能量低,呈反芳香性;当有奇数个电子对时,基态为L+R,呈芳香性。环丁二烯分子有偶数个电子对,故为反芳香性化合物。对于联环丁二烯分子,它们具有数个键交替的价键共振结构,可以形成多种in-phase或out-of-phase的线性组合波函数,如图4所示。它们均有偶数个共振电子对,基态为out-of-phase,即RR-LL或RR-RL等,体系应该呈反芳香性,价键理论的预测与实验相符。

图4 二环丁烯反芳香性的双态价键模型
除用于判断芳香性之外,价键理论在处理更复杂的化学体系时也具有明显优势,较为典型的例子是对Lin金属团簇的电子结构预测[25]。利用价键理论发现Li原子团簇除了以电子配对成键形式存在之外,还存在极高自旋态,且Lin的稳定化能随n的增加而增大。n=2时每原子的稳定化能只有2.5 kJ·mol-1,而n=7时则达到32.7 kJ·mol-1,最大可至50.2 kJ·mol-1。这是一种不通过电子配对方式形成的共价键,称为不成对铁磁性键。这种价键理论的计算结果是常规的分子轨道理论计算所无法比拟的,只有充分考虑组态相互作用之后,分子轨道理论才能得到与价键模型相一致的结论。
4 结语
价键理论与分子轨道理论自始至终一直存在各种争论。2003年Hoffmann、Shaik、Hiberty三人[26]曾对这两种量子力学理论的历史和发展进行了充分讨论。事实上VB和MO并不存在本质的区别,更不是相互对立的两种理论体系,二者的所谓“区别”只不过是由于所考虑的截断或近似不同而导致的表面现象。当严格求解时,无论价键理论还是分子轨道理论,都能够正确描述客观事实。虽然分子轨道理论更易于研究结构或者与轨道对称性有关的问题(如激发态),而价键理论在研究反应势垒、反应机理等方面具有优势。只有恰当地比较价键理论和分子轨道理论的优缺点及其应用范围,才能有效避免各种片面性的认识。
与此同时,我们发现追踪原始文献对辨析结构化学中的模糊概念起着至关重要的作用。因此,在结构化学的教学过程中,应强化学生对原始文献的认知,提高学生对原始文献的重视程度,这对深化研究型教学改革非常有利。
[1]潘道凯,赵成大,郑载兴.物质结构.第2版.北京:高等教育出版,1994.
[2]李炳瑞.结构化学.第1版.北京:高等教育出版社,2004.
[3]林梦海,林银钟.结构化学.北京:科学出版社,2004.
[4]Atkins,P.;Jones,L.Chemical Principles;W.H.Freeman and Company:New York,1999.
[5]周公度,段连运.结构化学基础.第4版.北京:北京大学出版社,2008:93.
[6]徐光宪,王祥云.物质结构.第2版.北京:高等教育出版社,1987:150.
[7]江元生.结构化学.第3版.北京:高等教育出版社,2004:89.
[8]Li,W.K.;Zhou,G.D.;WaiMak,T.C.Advanced Structural Inorganic Chemistry;Oxford University Press:New York,2008;pp 95.
[9]朱月香,钱民协,段连运.大学化学,2011,26(5),15.
[10]王腾,刘晶静,孙宏伟.大学化学,2015,20(3),14.
[11]Lewis,G.N.J.Am.Chem.Soc.1916,38(4),762.
[12]Heitler,W.;London,F.Z.Phys.1927,44,455.
[13]Wang,S.C.Phys.Rev.1928,31,579.
[14]Pauling,L.J.Am.Chem.Soc.1931,53,1367.
[15]Mulliken,R.S.Phys.Rev.1928,32,186.
[16]Mulliken,R.S.Phys.Rev.1928,32,213.
[17]Lewis,G.N.J.Am.Chem.Soc.1924,46,2027.
[18]Pauling,L.J.Am.Chem.Soc.1931,53,3225.
[19]Weizel,W.Z.Phys.1929,59,320.
[20]Goddard,W.A.;Dunning,T.H.;Hunt,W.J.;Hay,P.J.Acc.Chem.Res.1973,6,368.
[21]McWeeny,R.J.Mol.Struct.(THEOCHEM)1991,229,29.
[22]Huckel,E.Z.Phys.1930,60,423.
[23]Craig,D.P.J.Chem.Soc.1951,3175.
[24]Zilberg,S.;Haas,Y.J.Phys.Chem.A 1998,102,10843.
[25]Danovich,D.;Wu,W.;Shaik,S.J.Am.Chem.Soc.1999,121,3165.
[26]Hoffman,R.;Shaik,S.;Hiberty,P.C.Acc.Chem.Res.2003,36,750.
Revisit on the Teaching of Valence Bond Theory in the Curriculum of Structural Chemistry
HOU HuaHUANG Zhi-ChaoWANG Bao-Shan*
(College of Chemistry and Molecular Sciences,Wuhan University,Wuhan 430072,P.R.China)
After extensive investigations on the valence bond theory in the curriculum of structural chemistry,it was found that the limitation or even failure of the valence bond theory especially for the double bond electronic structure of oxygen molecule does not exist.Valence theory is capable of not only rationalizing or predicting the molecular structures but also revealing the chemical nature for many properties such as aromaticity and ferromagneticity in comparison with molecular orbital theory.
Valence bond theory;Chemical bond;Oxygen;Aromaticity
O6;G64
10.3866/PKU.DXHX201512025
,Email:baoshan@whu.edu.cn
武汉大学教改项目(QYW201206)
猜你喜欢
军事文摘(2023年22期)2023-12-19 06:41:04
中学生数理化·中考版(2022年9期)2022-10-25 03:49:04
数学物理学报(2022年3期)2022-05-25 13:33:22
数学物理学报(2022年1期)2022-03-16 06:15:04
中学生数理化·中考版(2021年9期)2021-11-20 06:17:38
数学物理学报(2021年5期)2021-11-19 07:01:16
大学化学(2021年8期)2021-09-26 10:50:46
数学物理学报(2021年3期)2021-07-19 06:02:18
山东化工(2020年5期)2020-04-07 09:59:30
儿童故事画报·自然探秘(2016年6期)2016-09-14 02:08:56
