
Fe(PDP)催化的C-H键活化
2016-11-01 06:20:12周小洲
大学化学 2016年9期
周小洲
(北京大学化学与分子工程学院,北京 100871)
·未来化学家·
Fe(PDP)催化的C-H键活化
周小洲*
(北京大学化学与分子工程学院,北京100871)
介绍高效、高选择性的Fe(PDP)催化体系的功能,并总结其选择性规律。将此规律应用在复杂分子体系中依然可行。根据反应的选择性特点和金属催化活化碳氢键的研究结果,提出了一种机理假设。最后,讨论了Fe(PDP)体系今后的可能发展方向。
碳氢键活化;Fe(PDP);金属催化剂;机理
碳氢键是有机分子中最常见的化学键,同时也是长期以来公认的惰性化学键,该位点的转化通常需要多步反应才可实现;然而又因碳氢键数量多,所以这些反应、转化普遍缺乏选择性。生物体内的酶可以很好地完成这项工作,但其利用次级键配位环境选择底物分子的行为模式注定了其在普适性方面的缺陷,所以这个思路或许并不利于这方面研究的展开[1]。因此如何简捷且精确地活化碳氢键成了有机化学领域的一大热点。从20世纪开始,已陆续出现关于碳氢活化催化剂的报道,但这些催化体系多数不具有高选择性,催化效率也不理想,甚至有些还需要以被初步活化过的碳氢键为底物。这里将介绍一种高效、高选择性的Fe催化剂,它能够较好地完成碳氢键的活化,并一定程度地解决上述难题。
1 Fe(PDP)催化体系的性质、选择性及其应用
Sharpless[2]于1994年即已提出,可以采用大位阻的亲电金属配合物作为催化剂活化碳氢键,令其选择性地进行氧化、氨化或烷基化。顺此思路,先后出现了很多可活化碳氢键的催化剂[3]。在碳氢键氧化反应中,发现一些Fe配合物可在AcOH存在的条件下催化H2O2选择性氧化碳氢键的反应。以此为基础,经过配体优化后的Fe(PDP)(1)明显具有了更高的催化效率(图1),使其成为少数高效、高选择性的碳氢活化催化剂之一[4]。

图1 Fe(PDP)结构
在筛选出最优催化条件后,White和Chen[5,6]用小分子底物进行了一系列实验,并总结出了Fe(PDP)的催化选择性规律。他们首先讨论了底物电子分布对催化选择性的影响(图2)。由于Fe(PDP)本身为Lewis酸,有亲电性质,故推测在Fe(PDP)催化下,富电子环境下的碳氢键会优先反应。一系列实验表明,当存在如羰基、酯基或卤素等吸电子基团(EWG)时,无论底物是环烃还是链烃,通常只有最远离EWG的碳氢键会被H2O2氧化,选择率通常可达到50%以上。这一现象与之前的猜想一致,电子诱导效应对反应位点选择有显著影响。值得注意的是,反应中对于距离EWG等同距离的位点,三级碳相比于二级碳上的碳氢键有明显反应优势。从电子诱导效应的角度来看,这说明了Fe(PDP)对细微的电子分布差异可做出敏感的区别,这对精确的催化选择性意义重大。
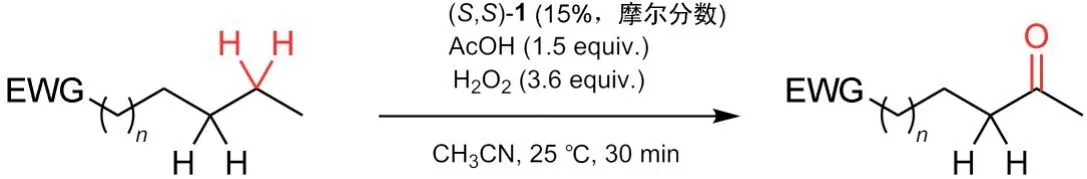
图2 最优条件下的电荷诱导效应
确定电子分布对反应的影响之后,他们又讨论了位阻对催化选择性的影响(表1)。由于Fe(PDP)是具有大位阻的配合物,故认为在其参与的催化过程中位阻小的位点优先反应。实验结果与推论一致:1,1-二甲基环己烷(#1)的氧化中C3上的氧化产物为主要产物,而非电荷分布上有利的C2;cis-1,2-二甲基环己烷(#2)的氧化中C1氧化产物为主产物;而trans-1,2-二甲基环己烷(#3)的氧化中则是C4氧化产物为主产物。这说明存在位阻区别时,基于电子诱导效应的选择规律可以被颠倒:考虑位阻区别时二级碳上的碳氢键可能会优先被氧化。

表1 最优条件下的空间位阻效应影响
由于Fe(PDP)本身为Lewis酸,强给电子基团的Lewis碱反应底物通常不适合此体系(如氧等碱性位点可能会直接与催化中心作用,从而阻止其与目标碳氢位点反应);但对于具有某些结构特征的该类分子,Fe(PDP)依旧能够通过一系列电子立体效应活化特定位点的碳氢键,达到选择性氧化的结果(表2)。如含有环丙烷结构单元的底物(#1),由于其键中含p轨道的π键特征,故可以通过超共轭效应活化本应被位阻效应屏蔽的α位点;又如环状底物(#2-#5)在此体系中进行氧化时,考虑到反应位点碳由sp3结构氧化后变成sp2结构可一定程度释放环张力,故有时也会形成并不完全符合上述两大判断依据的产物。随着环中碳数的增加,环的柔性增大,在大于六元环的体系中该氧化不会停止于羰基产物,而是会进一步氧化开环,最终生成二酸(#5)。
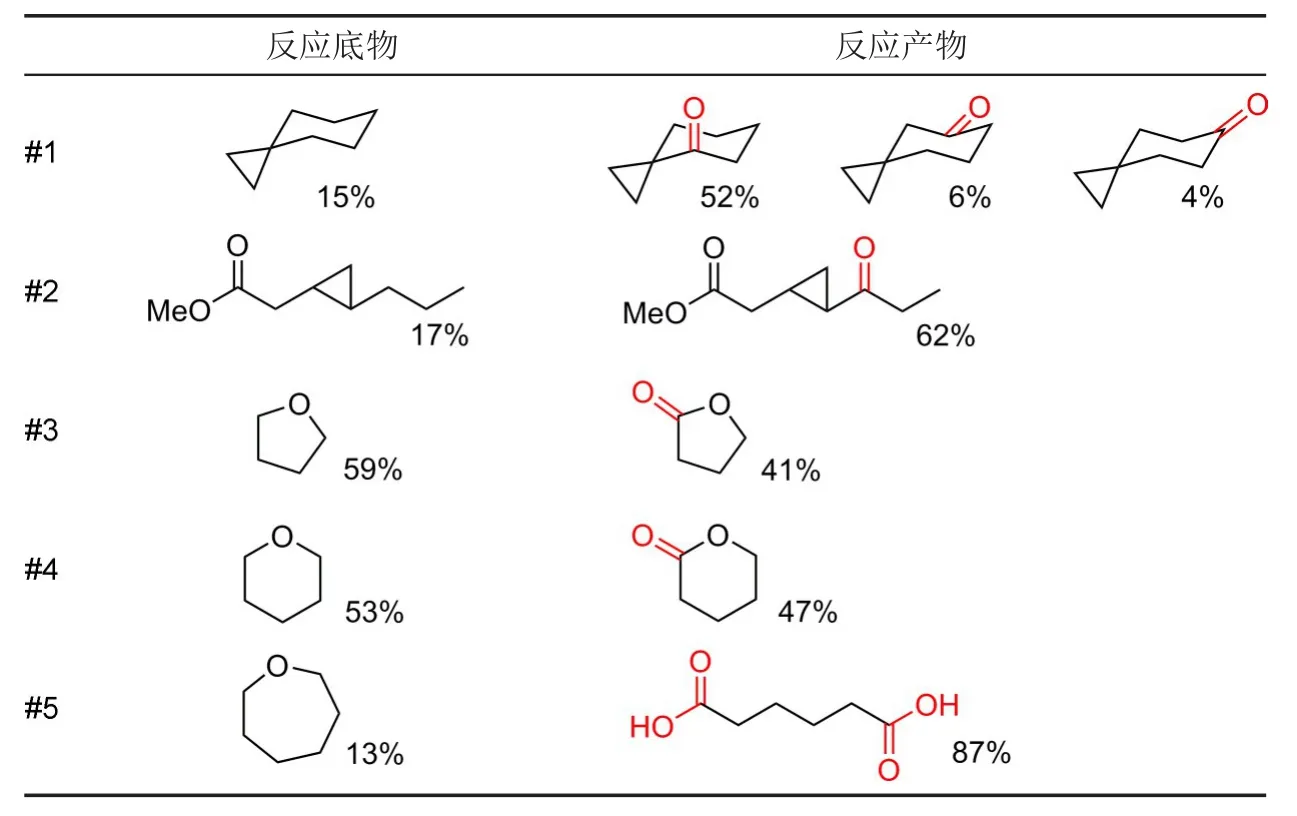
表2 最优条件下的电荷立体效应影响
White等在以结构较为简单的小分子作为模型建立Fe(PDP)选择性基本规律后,尝试将其应用在复杂分子的合成中,希望能够通过这些规律预测碳氢键氧化的反应位点。(-)-ambroxide通过分步氧化转换为(+)-2-oxo-sclareolide的反应就是验证上述规律在复杂分子体系中依然适用的很好的例子(图3)。(-)-ambroxide分子中,首先由于C环上氧原子的给电子能力使其邻位C-H键得到活化,故第一步氧化发生在这个位点,生成80%(+)-sclareolide。然后进一步考虑电子诱导效应,即第二步氧化应发生在尽可能远离EWG(此底物中的内酯)的位点上,故最可能反应的是A环上的亚甲基;结合空间位阻效应,即可确定氧代位置,最后得到46%(+)-2-oxo-sclareolide。由此可见,电子效应、位阻效应及其他电子立体效应的基本预测规律可以应用于复杂分子体系。

图3 (-)-ambroxide通过分步氧化生成(+)-2-oxo-sclareolide
2 Fe(PDP)催化氧化碳氢键的机理假设
早在1991年,McDouall等[7]就建立了通过亲电反应进行碳氢键氧化的理论模型,并结合分子轨道理论给出了机理假设。他们认为sp3碳的分子轨道首先与亲电试剂的空轨道结合,随后该碳上的氢发生迁移,与亲电试剂中原有电子对占据的轨道重新组合(图4)。由于他们用于建立模型的亲电试剂为过氧化物,故反应至此完成。1999年,Que和Chen[8]提出了在烷烃催化氧化过程中有高价金属氧代物参与的机理假设,并通过同位素标记等方法得到了金属氧代物形成及其后续转化的证据。
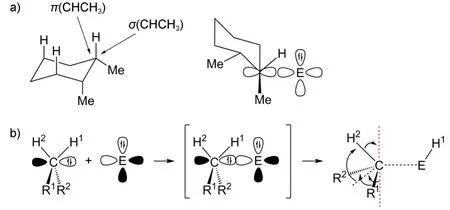
图4 (a)碳氢键氧化机理模型;(b)由假设机理解释cis-1,2-二甲基环己烷氧化选择性
结合上述研究成果,这里笔者提出了一种Fe(PDP)催化氧化碳氢键反应的机理假设(图5)。Fe(PDP)首先脱掉两个乙腈配体暴露出Fe的配位位点,然后与氧化剂H2O2反应生成氧代高价金属配合物;此时底物C-H分子轨道与金属中心空轨道作用成键,通过四元环过渡态发生氢迁移,生成中间物种;最后通过还原消除得到目标产物,释放活性催化剂,完成催化循环。
这一机理可解释Fe(PDP)催化中影响选择性的电子诱导效应和空间位阻效应:整个催化循环的关键在于C-H分子轨道中的成键电子对Fe中心的配位,故反应位点的电荷密度是影响反应的重要因素;分子轨道组合要求轨道对称性匹配、方向正确,故限制了底物分子与催化剂的作用方向,一定程度加大了位阻影响。上文中提及的cis-1,2-二甲基环己烷一例可由此解释(表1#2)。实验中三级碳位点被氧化后,其立体化学的完全保留也从另一个侧面验证了这一机理的合理性。其中配体的主要作用是提供离域π体系,稳定中间物种的四价Fe金属中心,同时提供不易变形的大位阻保证空间选择性。此机理是否合理还有待进一步实验证明。
3 Fe(PDP)催化体系的完善方向
Fe(PDP)体系是目前少有的能够较为准确预测碳氢键选择性氧化位点的催化剂体系,但从本质上讲依旧只能针对传统意义上最有活性的位点进行氧化,其选择性也仍依赖于底物分子中各个碳氢键的自身性质。理想的碳氢键活化催化剂的选择性,应该做到主动从催化剂的修饰出发,选择所需位点进行反应。简而言之,是针对所需位点修饰催化剂,而非因为催化剂有某种特性而只使用它做某种位点的反应。Fe(PDP)这类催化剂具有向理想化催化体系发展的潜质。这是由于多数碳氢键活化催化剂(如贵金属等)均是以某一种效应为影响因素,由于只有一个尺度方向能够调整,所以这样的催化剂很难针对活性适中的位点进行活化;而Fe(PDP)体系则不同,除去由于不同分子的不同结构造成的电子立体效应外,还有电子诱导和位阻两个尺度方向可以调控,这样一来便可以反过来通过规律预测怎样修饰的催化剂能够活化何种特定位点的碳氢键。例如,现有一复杂分子需要实现某一个特定位点的碳氢键氧化,其位置从电子诱导效应的角度来看活性较低,但从位阻角度来看却利于反应。此时可以考虑在不显著改变催化剂电子分布的同时增大配体位阻,以达到位阻效应控制的目的(Fe(PDP)催化中二级碳的碳氢键优先反应即如此类);反之则可以考虑在不改变配体位阻的情况下增强催化剂的Lewis酸性来实现优化。如此应用选择性规律,则可根据复杂分子合成中的实际需求,对已有的催化剂骨架进行修饰。
图5 提出的机理假设
Fe(PDP)在活化碳氢键的反应中优势突出:其具有高催化效率和高选择性,且整个催化体系符合环境友好的要求。在最优条件下,该催化剂的选择性可以通过分析底物分子的电子分布、位阻及电子立体效应进行较为精确的预测。Fe(PDP)体系通过合理修饰,有发展为理想选择性的催化体系的可能性。这里还提出了Fe(PDP)催化过程的假设机理,但其正确性还有待进一步实验验证。
[1]Clardy,J.;Walsh,C.Nature 2004,432,829.
[2]Sharpless,K.B.Tetrahedron 1994,65,4235.
[3]White,M.C.Science 2012,335,807.
[4]England,J.;Doyle,G.J.;Jacobsen,E.N.J.Am.Chem.Soc.2001,123,7194.
[5]Chen,M.S.;White,M.C.Science 2007,318,783.
[6]Chen,M.S.;White,M.C.Science 2010,327,566.
[7]Bach,R.D.;Andres,J.L.;Su,M.D.;McDouall,J.J.W.J.Am.Chem.Soc.1993,115,5768.
[8]Chen,K.;Que,L.,Jr.Chem.Commun.1999,1375.
[9]Yu,J.Q.;Shi,Z.C-H Activation;Springer:Heidelberg,2010.
[10]Kűrti,L.;Czakó,B.Strategic Applications of Named Reactions in Organic Synthesis;ELSEVIER:San Diego,2005.
Activation of C-H Bonds Catalyzed by Fe(PDP)Catalytic System
ZHOU Xiao-Zhou*
(College of Chemistry and Molecular Engineering,Peking University,Beijing 100871,P.R.China)
The Fe(PDP)catalytic system which processes high efficiency and selectivity is introduced in this paper.Its selectivity rules have been clarified and applied to complicated organic molecule system. Based on the selectivity features of Fe(PDP)and current research advances on activation of C-H bonds by metal catalysis,a hypothetical mechanism is proposed.
C-HActivation;Fe(PDP);Metal catalyst;Mechanism
O6;G64
10.3866/PKU.DXHX201601019
,Email:annezhou@pku.edu.cn
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
云南化工(2021年6期)2021-12-21 07:30:56
科学(2020年2期)2020-08-24 07:57:00
第一财经(2019年8期)2019-08-26 17:53:46
哈尔滨医药(2015年2期)2015-12-01 03:57:13
学习月刊(2015年14期)2015-07-09 03:37:48
生物技术通报(2015年1期)2015-04-10 16:15:19
物理化学学报(2015年5期)2015-02-28 17:34:47
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年1期)2014-02-28 17:30:06
