
基于液相色谱-串联质谱法检测多种食品中的9种非食用色素
2016-09-26 06:40:10高莹高杰张勋董群孙岩付瑶赵韫慧邢燕燕
食品与发酵工业 2016年1期
高莹,高杰,张勋,董群*,孙岩,付瑶,赵韫慧,邢燕燕
1(长春理工大学 化学与环境工程学院 吉林 长春,130022) 2(吉林出入境检验检疫局 吉林 长春,130062)
基于液相色谱-串联质谱法检测多种食品中的9种非食用色素
高莹1,高杰1,张勋2,董群1*,孙岩2,付瑶2,赵韫慧2,邢燕燕2
1(长春理工大学 化学与环境工程学院 吉林 长春,130022) 2(吉林出入境检验检疫局 吉林 长春,130062)
建立了多种食品(葡萄酒、乳饮料、调制乳、海虾,熟鸡蛋,咸鸭蛋,肉制品,辣椒酱,辣椒粉)中的9种非食用色素酸性红1、酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B的液相色谱-串联质谱定量检测方法。分别对提取方式、不同食品的分类预处理方法和质谱条件进行了优化。并选择离子检测进行阳性验证,外标法定量分析了上述食品样品中的色素含量。该方法对于酸性红的最低检出限、线性范围和方法回收率分别为:0.10mg/kg、0.10~0.50mg/L和86.5%~95.3%;对于其他8种非食用色素的最低检出限、线性范围和方法回收率分别为:0.010mg/kg、0.010~0.10mg/L和86.5%~95.7%。
非食用色素;分类预处理方法;液相色谱-串联质谱法
色素(着色剂)作为食品添加剂可分为2种:(1)食品天然色素;(2)人工合成色素。天然色素较为安全,是从天然的动植物、微生物中提取得到,其色泽自然但生产成本高,化学性质不稳定;人工合成色素是用人工化学合成方法所制得的有机色素,本身无营养价值。与天然色素相比较,人工合成色素更能增加食品外观美感且能提升人的购买欲和食欲,而且价格低廉,色彩亮丽,性质稳定,配色方便易溶解,易调色。因此人工合成色素已广泛应用于食品工业[1-3]。但是在人工合成着色剂中,工业色素(酸性红1、酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙和罗丹明B等)不允许在食品中添加。因为这些工业色素的合成多以苯、甲苯、萘等芳烃类化工产品为原料,经过磺化、硝化、偶氮化等一系列有机反应化合而成,且多数为偶氮化合物,在机体内经过生物转化,可形成致癌物。同时在合成过程中产生的杂质如As、Hg、苯酚、苯胺、Pb、Cr、乙醚、氯化物、硫酸盐等均有不同程度的毒性,严重影响消费者的健康。当前,有一些不法商贩因抵挡不住利润的诱惑,在生产的食品中违法添加一些廉价的工业色素。自 2005 年 2 月,英国食品局发现有 350 多种食品受“苏丹红I号”污染之后,一种叫做“对位红”的色素又被发现添加于35种食品中。同时,我国国内也发现有不法商贩为使鸭蛋、辣椒酱具有诱人的色泽,在其中添加苏丹红,还有用“碱性玫瑰精(罗丹明B)”为海虾染色和用“碱性橙31”、“酸性橙7”和“间胺黄”在豆制品上染色。这些工业合成染料,可在人体的代谢分解过程中产生可致癌的芳香胺类物质,对广大消费者的身体健康造成严重危害。因此,建立快速、有效的食品中非法添加物的检测方法,有效地控制工业染料在食品中的滥用,也可为应对突发食品安全事件提供理论依据,具有重要的社会意义[4-7]。
目前,关于碱性橙31、罗丹明B、酸性橙7和酸性红Ⅰ的检测方法已有报道,但同时测定几种染料的方法却很少。随着食品安全问题日益严峻,有必要对食品中的禁用色素检测方法做出优化[8-11]。本文讨论和建立了多种食品(葡萄酒、乳饮料、调制乳、海虾,熟鸡蛋,咸鸭蛋,肉制品,辣椒酱,辣椒粉)的分类预处理方法和这些食品中9种非食用色素的液相色谱-串联质谱定量检测方法。并且采用了阳性确证方法,针对选定的特征离子对9种人工合成色素进行了定性及定量分析。
1 实验部分
1.1主要仪器与试剂
API4000Q液相色谱-串联质谱仪(配有电喷雾离子源(ESI)的两级串联四级杆质量选择检测器),美国AB公司;涡旋振荡器,器美国Scientificindustries公司;AS20500A超声波清洗器,中国天津AUTOScience公司;天平:感量为0.1mg和0.01g,德国梅特勒公司;高速冷冻离心机,美国贝克曼库尔特公司,最大转速4 000r/min;氮气浓缩仪,美国Zymark公司;涡旋混合器,美国Scientificindustries公司;聚丙烯离心管:50mL,具塞;移液器:1 000、100μL。
甲醇、甲酸、乙腈、正己烷、乙醇,均为色谱纯;NaCl;分析纯;0.1%甲酸水溶液(1mL甲酸溶解于1 000mL水中);90%乙腈水溶液(量取900mL乙腈,100mL水,混匀,使用前配制);标准物质:酸性红1(azophloxine,CASNO. 3734-67-6,C18H13N3NaO8S2)、酸性橙7(orange,CASNO. 633-96-5,C16H11N2NaO4S)、酸性黄36(metanilyellow,CASNO. 587-98-4,C18H14N3NaO3S)、苏丹红Ⅰ(sudanI,CASNO. 842-07-9,C16H12N2O)、苏丹红Ⅱ(sudanII,CASNO. 3118-97-6,C18H16N2O)、苏丹红Ⅲ(sudanⅢ,CASNO. 85-86-9,C22H16N4O)、苏丹红Ⅳ(sudanⅣ,CASNO. 85-83-6,C24H20N4O)、碱性橙31(basicorange31,CASNO. 97404-02-9,C11H14ClN5)、罗丹明B(basicrosered,CASNO. 81-88-9,C28H31ClN2O3),纯度均≥97%,国药集团化学试剂有限公司;标准储备溶液:准确称取适量的标准品,用水配制成1.0mg/mL的标准贮备液,在0~4 ℃冰箱中保存;标准工作溶液:根据需要用水稀释配制适当浓度的标准工作液,现用现配;微孔滤膜:有机系,0.20μm。
1.2液相色谱与质谱条件
液相色谱条件:色谱柱:WatersXBridgeC18,250mm×4.6mm,粒度5.0μm或相当者;柱温:30 ℃;进样量:20μL;流动相:A:0.1%甲酸水,B:乙腈,C:水;梯度:t=0min, 10%B,90%C;t=6.0min时,10%A,90%B;t=17.1min时,90%B,10%C;流速:0.30mL/min。
质谱条件:离子源,电喷雾TurboSpray,正、负离子扫描;气路参数,CUR,25.00Psi;GS1,65.00Psi;GS2,55.00Psi;IS,-4 500.00V;TEM,550.00 ℃;CAD,8.00Psi;扫描方式,多反应监测(MRM)。
负离子质谱条件:气帘气(CUR),13Psi;雾化气(GS1),20Psi;辅助气(GS2),20Psi;电喷雾电压(IS),-4 500.00V;碰撞气(CAD),6.0Psi;离子源温度(TEM),550.0 ℃;
正离子质谱条件:气帘气(CUR),13Psi;雾化气(GS1),20Psi;辅助气(GS2),20Psi;电喷雾电压(IS),4 500.00V;碰撞气(CAD),6.0Psi;离子源温度(TEM),550.0 ℃。
1.3样品预处理,提取与纯化
本试验中,实测的样品有9种,分别为葡萄酒、乳饮料、调制乳、海虾,熟鸡蛋,咸鸭蛋,肉制品,辣椒酱和辣椒粉,它们相对应的预处理、提取和净化步骤如下:
(1)葡萄酒:取样品1kg充分混匀,气泡酒先超声脱气,再将混匀样品装在洁净容器内密封,作为试样,置于0~4 ℃保存。移取10mL试样于50mL聚四氟乙烯离心管内,加入5gNaCl、20mL乙腈,涡旋混匀1min,超声提取10min,10 000r/min离心5min,上层全部提取液转移至旋蒸瓶中,残渣再加入20mL乙腈,重复上述操作,合并2次提取液,38 ℃水浴旋蒸至尽干,用V(乙腈)∶V(水)=9∶1溶解,定容至10mL,取1mL过0.20μm滤膜,供液相色谱-质谱/质谱仪测定。
(2)乳饮料、调制乳:准确称取10mL样品于50mL聚四氟乙烯离心管内,加入15mL乙腈,涡旋混匀1min,超声提取10min,10 000r/min离心5min,上层全部提取液转移至旋蒸瓶中,残渣再加入15mL乙腈,重复上述操作,合并2次提取液,剩下操作同上述葡萄酒样品的处理方法。
(3)海虾、煮熟后的鸡蛋、咸鸭蛋、肉制品、辣椒酱、辣椒粉:将体积过大的样品绞碎,称取 10g(准确至 0.01g) 样品于50mL聚四氟乙烯离心管中,加入20mL正己烷,涡旋混匀1min,超声提取10min,10 000r/min离心5min,上层全部提取液转移至旋蒸瓶中,残渣再加入20mL正己烷,重复上述操作,合并2次提取液,剩下操作同上述葡萄酒样品的处理方法。
2 结果与讨论
2.1提取方法的选择
当前常用的提取方法有索氏提取法、漂洗法、振荡法、组织捣碎法、浸渍提取法、均质法、超声波提取法、消化法、柱层析洗脱法等。文献报道的提取方法一般采用振荡法和超声波提取法,本试验在10mL本底值为零的乳饮料中加入15mL乙腈,酸性红1添加0.10mg/kg,酸性橙7、酸性黄36,苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B分别添加0.010mg/kg水平的样品,分别用2种方法提取并进行回收测定。
由图1可知,2种提取方法提取效果相当,考虑到超声波提取法比振荡法提取时间更短,可提高检测工作效率,本实验选用超声波提取法进行多种食品中酸性红1、酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B这9种工业染料的提取。
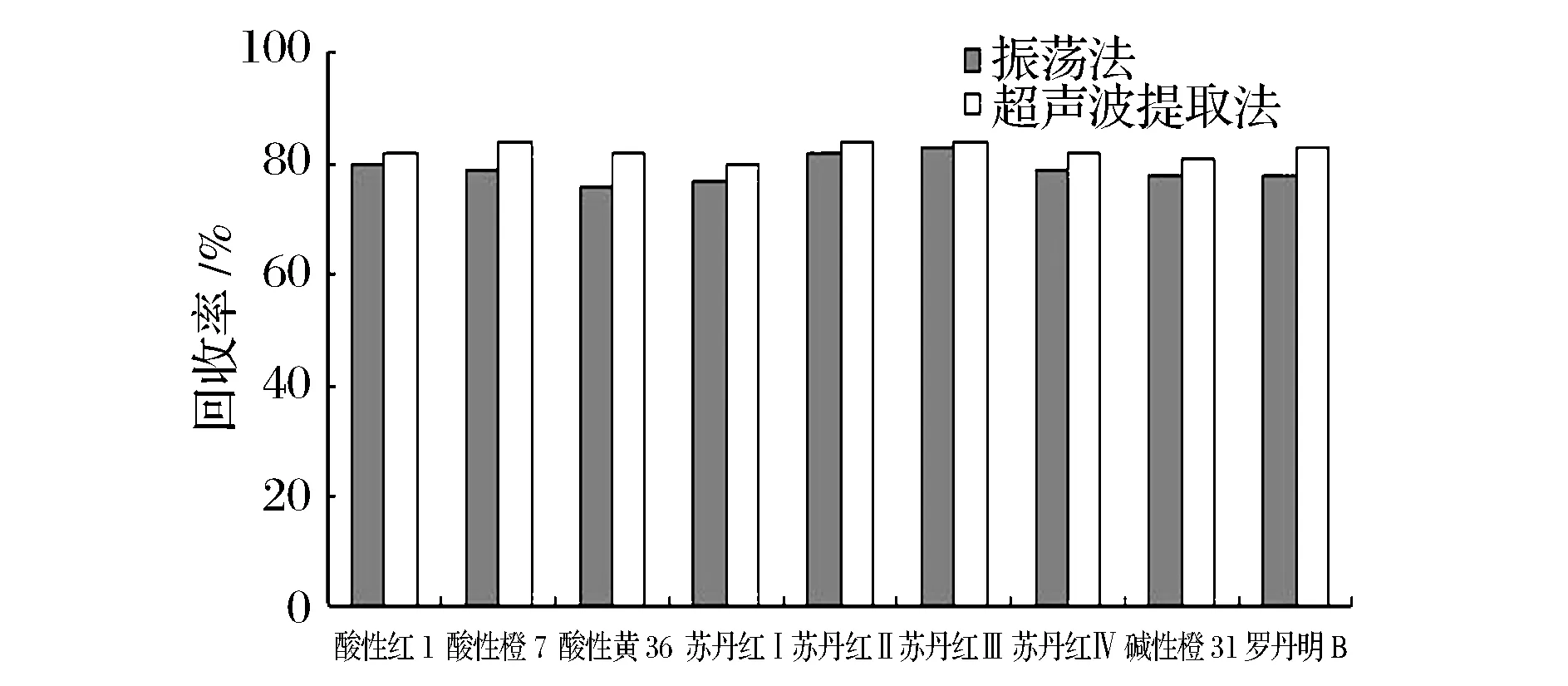
图1 提取方式对回收率的影响Fig.1 The impact of the extraction method on recovery
2.2提取溶剂的选择
根据相似相溶原理,以试样的种类、待测物的性质和提取溶剂的性质3个方面进行综合考虑,以单一溶剂或混合溶剂进行提取是确定选用提取溶剂的原则。本课题研究的9种非食用色素在有机溶剂中溶解性较好,因此,应根据不同的食品种类选用合适的有机溶剂兼顾这9种非食用色素的检测需要。
对于含有较多蛋白质的液体食品,例如乳饮料,调制乳等,为了提高提取率,先将蛋白质分离后再进行测定。根据文献可知,乙腈具有快速沉淀蛋白的作用,能够使目标化合物与基体干扰物分离,达到分离和净化的目的,因此对于含有较多蛋白质的液体食品选用乙腈进行提取。为了确定所用提取液乙腈的体积,本试验在10mL本底值为零的乳饮料中加入10、15、20mL乙腈,酸性红1添加0.10mg/kg,酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙、罗丹明B分别添加0.010mg/kg进行回收测定。由图2可知,采用15mL乙腈沉淀蛋白的效果最佳,用V(乙腈)∶V(水)=9∶1溶解后得到的溶液较澄清且色素回收率较高,因此在提取蛋白质含量较高的液体食品时,所用乙腈的体积为15mL。对于含有CO2的液体食品,例如碳酸饮料,啤酒,果汁饮料等,为了避免CO2对提取的影响,应该先进行超声脱气(除去CO2),然后再用乙腈作为提取溶剂进行提取。
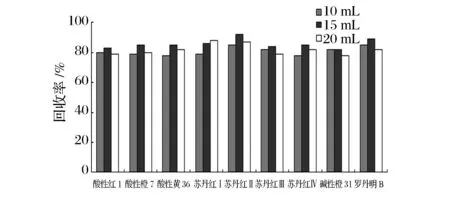
图2 乙腈体积对回收率的影响Fig.2 The impact of the volume of acetonitrile on recovery
对于含有较多蛋白质、脂肪的固体食品,如海虾,煮熟后的鸡蛋,咸鸭蛋,肉制品,辣椒酱,辣椒粉等,对提取溶剂的选择进行了摸索性实验。本试验以本底值为零的肉制品为例进行了乙醇,乙腈和正己烷提取的效果实验,称取10g(准确至 0.01g)样品3份于50mL聚四氟乙烯离心管中,酸性红1添加0.10mg/kg,酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B分别添加0.010mg/kg水平的样品。分别加入20mL乙醇,乙腈和正己烷提取。由图3可知,乙醇的提取效果尚可,但乙醇提取液中含水量较大,在旋转蒸发浓缩的操作过程中,很难旋至近干,且在减压浓缩时容易发生暴沸,造成仪器被污染和目标物的损失,可操作性不强。乙腈和正己烷作为提取剂的回收率均较好,与乙腈相比,正己烷的折射率比较低,与基体干扰物的作用也比较弱,能够有效的除去水溶性蛋白等杂质,用V(乙腈)∶(水)=9∶1溶解后得到的溶液较澄清,色素回收率高,加标回收率均高于80%,效果理想,达到了检测的要求,因此选用正己烷作为蛋白质、脂肪含量多的固体食品的提取溶剂。

图3 不同提取溶剂对回收率的影响Fig.3 The impact of the different solvents on recovery
2.3质谱条件的优化
以1μg/mL浓度的酸性红1、酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B9种工业染料标准溶液,针泵进样进入质谱系统。采用Q1全扫描(Q1MS)和碎片离子扫描(ProductIonMS2)方式分别确定母离子和子离子,并优化相关参数,表1为酸性红1、酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B9种工业染料的优化质谱条件。

表1 质谱优化条件
2.4质谱检测条件选择实验
通过针泵进样,进行Q1全扫描,选择合适的离子后,进行MRM多反应监测扫描,优化设定了碰撞气(CAD)、气帘气(CUR)、辅助气1(Gas1)、辅助气2(Gas2)、离子源温度(TEM)、去簇电压(DP)、入口电压(EP)、碰撞能量(CE)、出口电压(CXP)等参数。酸性红1(0.10mg/L)、酸性橙7(0.010mg/L)、酸性黄36(0.010mg/L)、苏丹红Ⅰ(0.010mg/L)、苏丹红Ⅱ(0.010mg/L)、苏丹红Ⅲ(0.010mg/L)、苏丹红Ⅳ(0.010mg/L)、碱性橙31(0.010mg/L)、罗丹明B(0.010mg/L)标准品的多反应监测(MRM)色谱图见图4。其中所测物质所对应的保留时间分别为:酸性红1(6.04min)、酸性橙7(7.26min)、酸性黄31(7.75min)、苏丹红Ⅰ(9.94min)、苏丹红Ⅱ(11.30min)、苏丹红Ⅲ(12.50min),苏丹红Ⅳ(14.72min)、碱性橙31(6.26min)、罗丹明B(8.01min)。
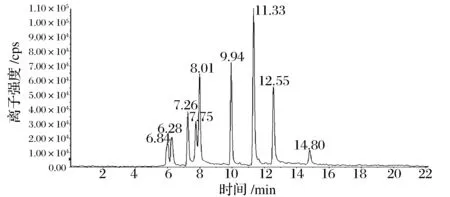
图4 标准溶液多反应监测色谱图Fig.4 MRM chromatogram of standard solution
2.5检出限和线性范围
在本方法所确定的LC-MS/MS检测条件下,酸性红1标准物在0.10~0.50mg/L,酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B8种染料标准物在0.010~0.10mg/L内制备5个浓度水平的标准溶液进行测定,每种标准物质在该浓度范围内呈良好线性关系,结果见表2。
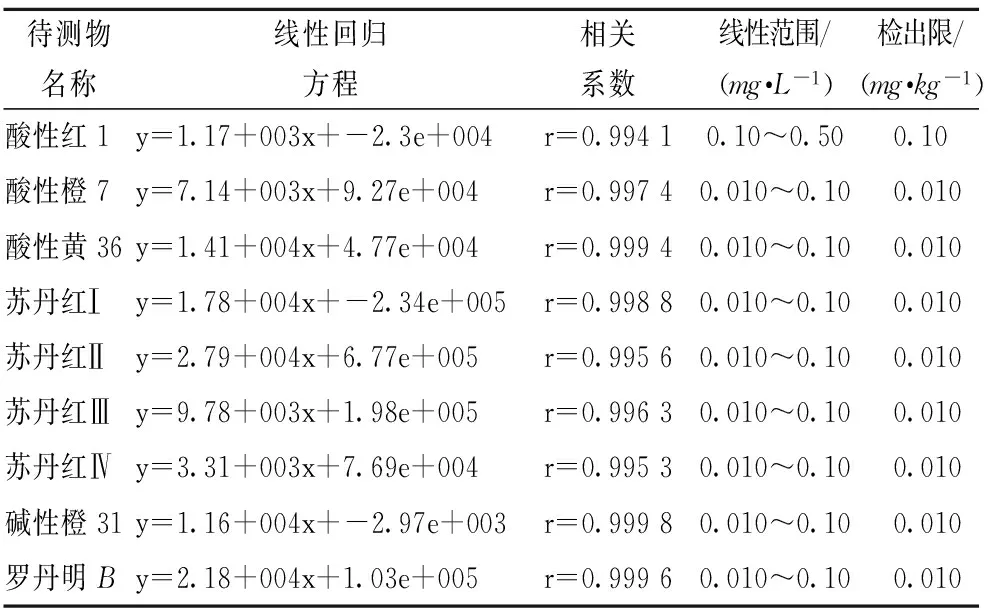
表2 方法的线性范围和检出限
2.6方法的精密度和回收率
采用添加法,即选择不含本底色素的食品,如肉制品,酸性红1添加0.10、 0.20、0.50mg/L,酸性橙7、酸性黄36、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、碱性橙31、罗丹明B分别添加分0.010、0.020、0.10mg/L水平的样品进行回收测定,每水平单独测定10次,回收率和精密度试验结果详见表3。经试验结果表明,平均添加回收率在86.5%~95.7%,相对标准偏差在5.7以内,测定结果令人满意。

表3 方法的精密度和回收率
续表3

色素名称本底值/(mg·kg-1)加标量/(mg·kg-1)测得平均值/(mg·kg-1)平均回收率/%RSD/%00.100.093293.21.9苏丹红Ⅰ00.0100.0089589.55.700.0200.0183691.92.300.100.093293.22.3苏丹红Ⅱ00.0100.0088588.53.100.0200.0180690.32.600.100.091391.32.2苏丹红Ⅲ00.0100.0086586.52.800.0200.0190295.13.300.100.091491.42.0苏丹红Ⅳ00.0100.0092592.52.500.0200.0184492.22.700.100.092892.82.4碱性橙00.0100.0093593.52.600.0200.018894.03.800.100.089289.25.700.0100.0091591.54.1罗丹明B00.0200.0191495.72.200.100.093193.11.4
2.7实际样品含量的测定
在该实验条件下,分别对10种不同样品进行了抽查测定,发现在白葡萄酒、乳饮料、海虾、咸鸭蛋、肉制品、辣椒酱、辣椒粉中不同程度的检出非食用色素,试验结果见表4。表4结果显示,不同种类样品中使用的非食用色素种类也不尽相同,在白葡萄酒中检出酸性橙7,乳饮料中检出酸性红1,海虾中检出罗丹明B,咸鸭蛋中检出苏丹红苏丹红Ⅰ,在肉制品中检出苏丹红Ⅱ、苏丹红Ⅳ,在辣椒酱、辣椒粉中检出苏丹红Ⅰ,上述结果证明该方法的研究和开发在食品安全的监管中是十分必要的和重要的。
3 结论
本实验建立了多种食品(葡萄酒、乳饮料、调制乳、海虾,熟鸡蛋,咸鸭蛋,肉制品,辣椒酱,辣椒粉)的分类预处理方法和这些食品中9种非食用色素的液相色谱-串联质谱定量检测方法。采用外标法定量、选择离子检测进行阳性确证,前处理简便、灵敏度高、准确性可靠、线性范围宽,适用范围广能够满足高质量检测的要求。
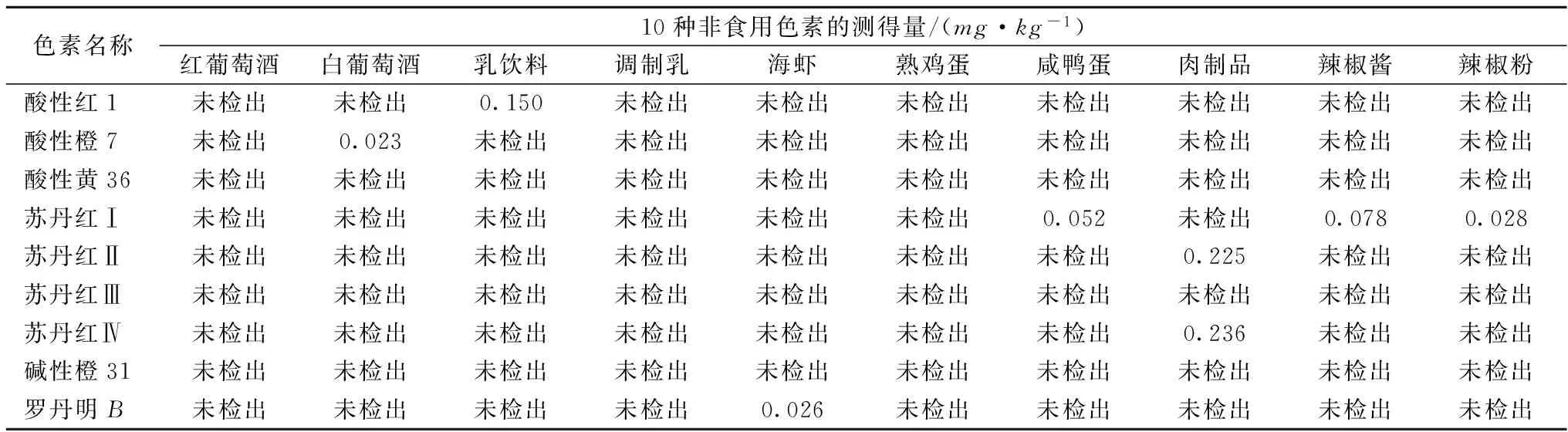
表4 实际样品的测定结果
[1]顾晓莉.HPLC测定食品中人工合成色素[J].吉林农业,2013,301 (2):78.
[2]卢士英,邹明强. 食品中常见的非食用色素的危害与检测[J].中国仪器仪表,2009(8):45-50.
[3]张春艳,卢华卫,郑书展,刘来俊,白国涛,李红. 调制乳中8种合成色素的快速测定[J].中国乳品工业,2011,39(10):48-53.
[4]曹丽芬,姚黎霞, 何良兴,茹巧美. 高效液相色谱法快速检测食品中苏丹红的含量[J]. 安徽农业科学,2014,42(2):1 532-1 533.
[5]沈磊. 高效液相-串联质谱测定食品中工业色素的研究[D].河北:河北科技大学,2013:1-2.
[6]ZDUNEKA,CYBULSKAJ,KONOPACKAD,etal.Newcontactacousticemissiondetectorfortextureevaluationofapples[J].JournalofFoodEngineering,2010,99(1):83-91.
[7]陈勇,花锦,潘亚利,王静慧,刘海峰. 超高效液相色谱-电喷雾串联质谱法同时测定调味品与熟肉制品中6种工业染料[J].中国调味品,2014,39(4):124-127.
[8]XUHui-rong,SUNBing.Variableselectioninvisibleandnearinfraredspectra:applicationtoon-linedeterminationofsugarconteneinpears[J].JournalofFoodEngineering,2012,109(1):142-147.
[9]LIAOQG,LIWH,LUOLG.Applicabilityofacceleratedsolventextractionforsyntheticcolorantsanalysisinmeatproductswithultrahighperformanceliquidchromatography-photodiodearraydetection[J].AnalChinaActa,2012,716(4):128-132.
[10]肖海龙,屠海云,王红青,等. 反相高效液相色谱法快速测定食品中18中水溶性合成着色剂[J].中国卫生检验杂志,2011,21(2):264-266.
[11]PETIGARAHB,MIRANDA-BERMUDEZE,BARROWJ.Determinationofsevencertifiedcoloradditivesinfoodproductsusingchromatography[J].JAgricFoodChem,2013,61(15):3 726-3 736.
Determinationof9illegalpigmentsinfoodbyLC-MS/MSmethod
GAOYing1,GAOJie1,ZHANGXun2,DONGQun1*,SUNYan2,FUYao2,ZHAOYun-hui2,XINGYan-yan2
1(CollegeofChemistryandEnvironmentalEngineeringChangchunUniversityofScienceandTechnology,Changchun130022,China) 2(JilinEntry-ExistInspectionandQuarantineBurean,Changchun130062,China)
Inthispaper,aquantitativemethodbasedonliquidchromatographytandemmassspectrometrywasestablishedtodetermine9illegalpigments(Azophloxine,Orange,Metanilyellow,SudanI,SudanII,SudanⅢ,SudanⅣ,BasicOrange31andBasicrosered)usedinvariouskindsoffoods(wine,milkbeverage,blendmilk,seashrimp,hard-boiledeggs,saltedduckeggs,meatproduct,hotpeppersauce,pepper,etc.).Thepre-processingmethodofdifferentkindsoffoods,theextractionmethodandthemassspectrumconditionsofvariouscolorantswereoptimized.ThenthecontentsofcolorantswerequantitativelydeterminedbyLC-MS/MSusingexternalstandardmethod.Thedetectionlimits,linearranges,andrecoveriesofthismethodforAzophloxine,were0.10mg/L, 0.10~0.50mg/Land86.5%~95.3%,respectively.Thedetectionlimits,linearranges,andrecoveriesfortheother8pigmentswere0.010mg/L,0.010 ~0.10mg/Land86.5~95.7%,respectively.
illegalediblepigment;classifiedpre-processingmethod;LC-MS/MS
10.13995/j.cnki.11-1802/ts.201601034
学士,副教授(董群助理研究员为通讯作者)。
国家质检总局科技项目(2012B134,2014IK135)
2015-04-16,改回日期:2015-12-22
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:28
食品安全导刊(2020年14期)2020-12-04 20:19:39
福建基础教育研究(2019年8期)2019-05-28 08:39:51
现代食品(2018年16期)2018-11-02 02:33:52
健康博览(2017年12期)2018-02-06 21:30:06
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:43
食品工业科技(2016年17期)2016-10-31 02:45:08
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
化工进展(2015年3期)2015-11-11 09:09:13
湖北理工学院学报(2015年1期)2015-02-27 15:02:28
