
煤焦油加氢脱硫精制研究进展
2016-09-26 08:33胡薇月崔文岗李稳宏
广州化工 2016年16期
胡薇月,李 珍,崔文岗,李 冬,李稳宏
(1 陕西省产品质量监督检验研究院,陕西 西安 710048;2 陕西省石油化工研究设计院,陕西 延安 710054;3 西北大学化工学院,陕西 西安 710069)
煤焦油加氢脱硫精制研究进展
胡薇月1,李珍2,崔文岗3,李冬3,李稳宏3
(1 陕西省产品质量监督检验研究院,陕西西安710048;2 陕西省石油化工研究设计院,陕西延安710054;3 西北大学化工学院,陕西西安710069)
综述了煤焦油原料中的含硫化合物的类型,主要包括硫醇、硫醚、二硫化物、噻吩、苯并噻吩、二苯并噻吩等含硫化合物。总结了不同类型含硫化合物在加氢脱硫过程中的反应机理,分析了煤焦油中典型含硫化合物加氢脱硫的反应动力学,并对其加氢脱硫的过程研究方向做了展望。旨在为煤焦油加氢脱硫精制过程提供一定的理论指导,避免煤焦油加氢精制催化剂研究和开发的盲目性
加氢脱硫;含硫化合物;反应机理;动力学
随着能源需求的不断增加,煤焦油的产量也呈逐年递增状态,煤焦油加氢制取清洁汽柴油的工艺也日趋成熟,此外,人们在不断探索新工艺的同时,也重视对已有制备过程中的缺陷进行改进。煤焦油中的硫化物会对各类金属设备造成严重腐蚀,;含硫化合物与氧化物、氯化物、氮化物、环烷酸和氢气等其它腐蚀性介质相互作用,形成多种含硫腐蚀环境,对炼油生产设备及发动机等有较大危害;各种含硫化合物燃烧之后均转化为SOx,对生态环境造成极大的危害。因此,为了解决油品存在的上述问题,必须对其进行加氢脱硫(HDS)精制处理,以使其达到清洁油品的标准。
本文主要对煤焦油中含硫化合物类型、加氢脱硫反应机理以及加氢脱硫反应动力学的研究进展进行了综述,并对未来的研究方向进行了展望。
1 煤焦油中含硫化合物的类型
硫在煤焦油原料中以元素硫、硫醇、硫醚、二硫化物等形态出现,从设备腐蚀与防护的角度考虑,一般将其分为活性硫和非活性硫。元素硫、硫化氢和低分子硫醇都能与金属直接作用而引起设备的腐蚀,因此它们统称为活性硫。其余不能与金属直接作用的含硫化合物统称为非活性硫,非活性硫在高温、高压和催化剂的作用下可部分分解为活性硫。
煤焦油中的含硫化合物有中性和酸性硫化物,包括硫醇、硫醚、二硫化物、噻吩、苯并噻吩(BT)、二苯并噻吩(DBT)、4-甲基二苯并噻吩(4-MDBT)和4,6-二甲基二苯并噻吩(4,6-DMDBT)等[1-5]。① 中性含硫化合物主要是具有噻吩环的化合物,其主要代表是噻吩、硫杂茚、硫芴和2,3-苯并硫芴,以及它们的甲基衍生物和少量的硫杂茚的二甲基衍生物。② 酸性含硫化合物主要是具有硫酚环的化合物,如苯硫酚、萘硫酚等,它们大部分属于高沸点化合物,主要存在于洗油馏分和蒽油馏分中。
2 加氢脱硫的反应机理
2.1非噻吩类硫化物的HDS反应
在加氢过程中,C-S键较易断开并生成相应的烃类和硫化氢,典型非噻吩类化合物的加氢脱硫反应如下:
(1)硫醇
(2)二硫化物
(3)硫醚
2.2噻吩类硫化物的HDS反应
2.2.1噻吩的HDS反应
噻吩的加氢脱硫反应如下:
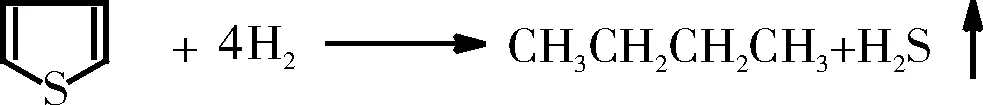
噻吩及其衍生物由于其中硫杂环的芳香性,难以氢解,导致噻吩硫要比非噻吩硫难脱除得多。噻吩的加氢脱硫反应是通过加氢和氢解两条平行的途径进行,由于硫化氢对C-S键氢解有强抑制作用而对加氢影响不大。因此,可以认为,加氢和氢解是在催化剂的不同活性中心上进行的。此外由于噻吩的结构特点和脱硫具有一定的难度,通常被用于含硫芳香类化合物HDS的模型。关于噻吩HDS的机理大多来自于微反装置[13-15]、金属单晶表面[16-18]、有机金属配合物[19]、担载的金属原子簇[20]等。对于文献中提出的噻吩HDS机理总结在图1中。
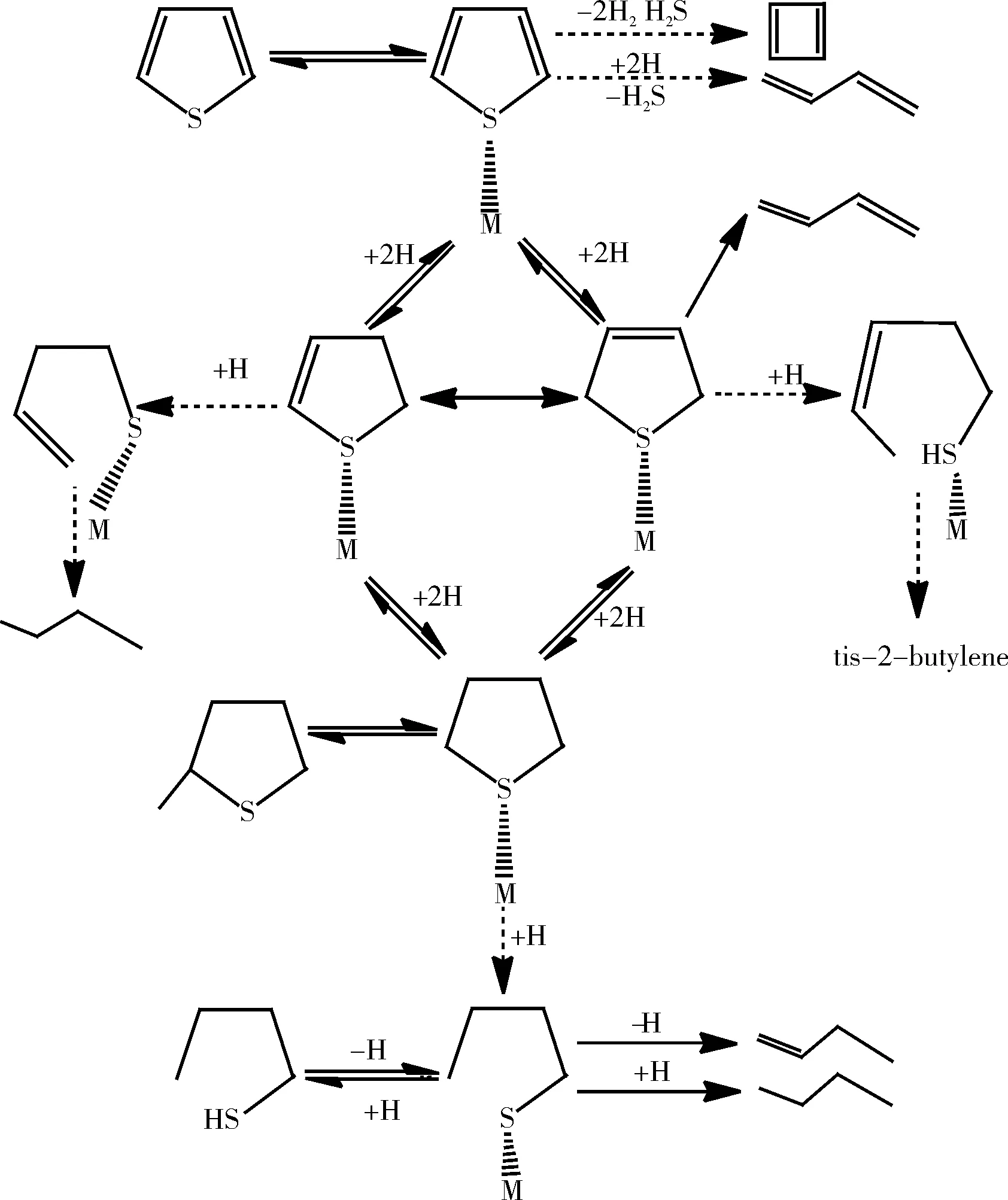
图1 噻吩HDS的反应途径
Lipsch[13]认为C-S键的断裂是首先经过氢解以形成丁二烯来完成的。Kolboe等[14]对噻吩、四氢噻吩(THT)和1-丁硫醇的HDS研究之后认为:噻吩和四氢噻吩没有经过加氢步骤,也不是经过相同的过渡态而脱硫。在HDS过程中,β-H向S原子转移,同时C-S断裂,即通过分子内氢解而完成。但是,更多的研究表明,要使芳环中的C-S直接断裂是相当困难的。对于噻吩的加氢脱硫,认为先进行C=C的饱和,然后再进行C-S的断裂[21]。由于加氢饱和破坏了噻吩环的芳香性,使得脱硫变得容易。在这一过程中二氢噻吩的氢解与C=C的加氢存在着竞争。但由于噻吩、二氢噻吩和四氢噻吩的HDS发现具有相同的产物分布,因而被认为是按照相同的机理进行的[22-24]。
Moser等[25]还提出了一种THT通过β位的H消去生成了丁烯硫醇盐的机理。部分加氢的观点认为,在相对低氢压下,部分加氢的噻吩生成了2,3-二氢噻吩和2,5-二氢噻吩中间体。Hesen等[26]通过负载在活性炭上的过渡金属硫化物催化剂催化噻吩HDS反应发现,加氢过程中有2,3-二氢噻吩生成,接着2,3-二氢噻吩通过脱硫、异构化生成2,5-二氢噻吩,或进行快速加氢作用生成THT。Hesen还指出,部分加氢中间体的产率与催化剂的活性反向相关,即HDS活性高时,中间体的产率很低,反之很高。
2.2.2苯并噻吩的HDS反应
苯并噻吩的加氢脱硫反应如下:
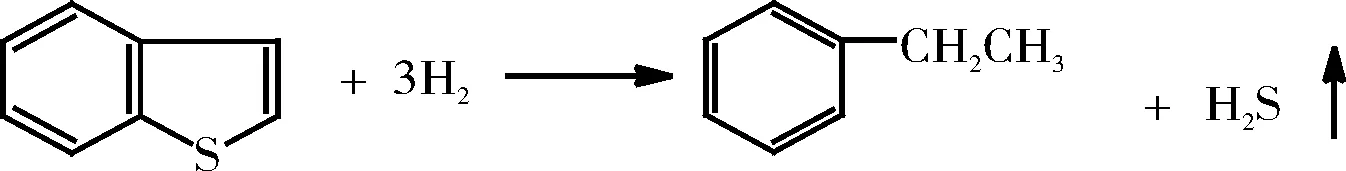
苯并噻吩在进行HDS时只生成乙苯和少量的二氢苯并噻吩[8-9],而二氢苯并噻吩进行HDS时,并没有观察到苯并噻吩的形成,所以可认为乙苯是通过二氢苯并噻吩作为中间物所形成的。Van Parijs等[28]提出了一个如图2所示的平行反应历程,与噻吩的HDS相似,也有直接脱硫和加氢脱硫两条途径。

图2 苯并噻吩的HDS的反应途径
2.2.3二苯并噻吩的HDS反应
二苯并噻吩的加氢脱硫反应如下:
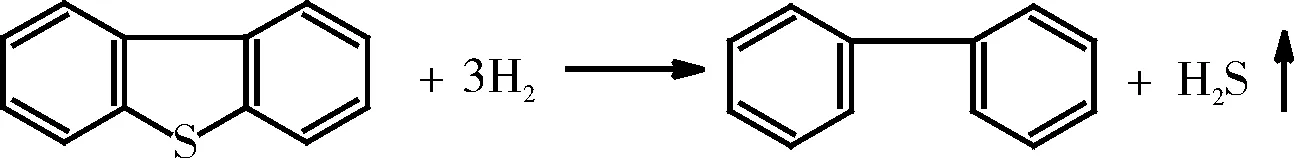
Gates等[29]对二苯并噻吩的HDS进行了研究,提出了图3所示的反应机理。因为发现联苯(Bi-Ph)是反应的主要产物,而环己基苯(CHB)只有极少量。在添加Bi-Ph和H2S的条件下进行二苯并噻吩的HDS时,发现Bi-Ph的添加显著地减少了二苯并噻吩的转化率,而添加H2S则没有影响,这表明二苯并噻吩的HDS受到了Bi-Ph的抑制[30]。
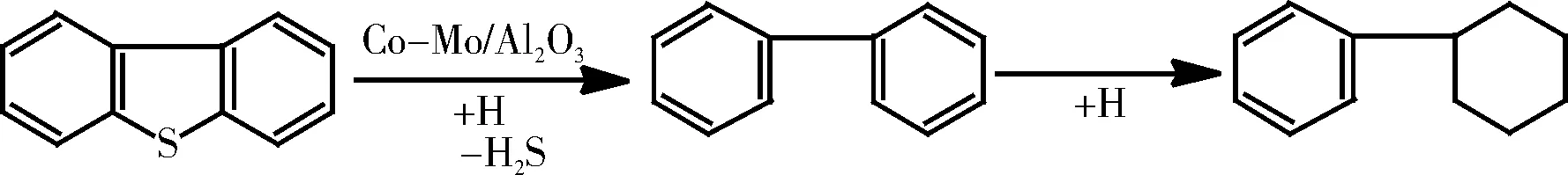
图3二苯并噻吩的HDS途径
Fig.3The pathway of dibenzothiophene HDS reaction
Rollman等[31]考察了二苯并噻吩的催化加氢,研究结果表明:环己基苯的量随温度的升高而增加,并认为环己基苯不是经由Bi-Ph的加氢所生成。Houalla等[35]提出了图4所示的反应机理,图中的数据表示在Co-Mo/Al2O3催化剂存在下,反应条件为30 ℃、10.2 MPa时的表观反应速率常数(L/g催化剂·s)。它表明反应可以通过最少量的氢耗来完成,且Bi-Ph与环己基苯的加氢速率很低。二苯并噻吩的加氢速率随着H2S的浓度的增加而增加,当采用Ni-Mo/Al2O3催化剂时得到的环己基苯的浓度比采用相似的Co-Mo/Al2O3催化剂高两倍。从这一反应历程看,主要反应是二苯并噻吩中C-S键断裂脱硫化氢生成联二苯,而芳香环先加氢再脱硫化氢的反应速率要慢得多,两者相差达600多倍。至于第二个苯环的加氢则更慢,所以主要产物是联二苯和环己基苯[36]。

图4 二苯并噻吩的HDS反应
3 加氢脱硫反应的动力学
HDS动力学研究不仅对研制新HDS催化剂的指导有重要意义,而且也是探讨各种硫化物在不同催化剂上的加氢反应机理的途径,还是开发新型加氢反应器和优化工艺的基础。
目前,有关HDS反应动力力学研究方法依据研究对象分为两种:(1)以真实油品作为研究对象,不具体研究某种物质的反应机理,而把反应过程当做黑箱模式,选择硫含量、密度、黏度和沸点等油品性质作为输入参数,脱硫率作为输出。考察反应条件以及性质参数对加氢脱硫结果影响规律,得到表观反应速率、脱硫率随参数和反应条件经验表达式,用于对研究对象脱硫率的预测以及反应条件的优化。不同研究者使用的催化剂、反应器、操作方式以及流体流动方式不完全相同,获得了很多具有特色和适用局限性的动力学模型,但目前为止没有一种动力学模型适用不同原料油品。(2)以模型化合物作为研究对象,对于深度脱硫,他们认为只要能够把含硫量较多且难脱除的二苯并噻吩类化合物中的硫脱除,就可以满足含硫量的要求,因此选择二苯并噻吩或4,6-二甲基-二苯并噻吩作为研究对象。这样可以把真实油品在催化剂上的复杂反应简单化,利于研究脱硫反应机理,分析影响脱硫的主要因素。
3.1噻吩
Satterfield和Roberts[42]根据稳态循环流动反应器的实验结果报道了噻吩的反应动力学结果,用Langmuir-Hinshelwood速率方程表示如下:
(1)
式中:k——速率常数
K——吸附平衡常数
P——分压
下标T——噻吩
下标H——氢
下标S——硫化氢
3.2苯并噻吩
Kilanowski和Gates[43]利用稳态微分流动反应器研究了苯并噻吩加氢脱硫反应动力学,催化剂为硫化的Co-Mo/Al2O3催化剂。在252 ℃、302 ℃和332 ℃的条件下粗略地可以用下列Langmur-Hinshelwood速率方程表示:
(2)
式中:KBT——苯并噻吩的吸附平衡常数
其他符号与式(1)中的含义相同。
3.3二苯并噻吩
根据Broderick[44]的研究报道,用硫化的Co-Mo/Al2O3催化剂在275~322 ℃,3.3~15.7 MPa的条件下得到的动力学方程如下:
(3)
式中:KDBT——二苯并噻吩的吸附平衡常数
Edvinsson等[45]研究二苯并噻吩的HDS动力学时认为:
(1)反应过程中存在两类活性位,即σ活性位和τ活性位;
(2)在σ活性位上进行直接脱硫反应,同时发生氢解反应;在τ活性位上二苯并噻吩先加氢部分饱和,随后再进行脱硫反应;
(3)反应速率较低,表面反应为速率控制步骤;
(4)反应速率对二苯并噻吩和H2均为1级反应;
(5)体系中H2S浓度很低,对反应的影响可以忽略。以此推导出了二苯并噻吩在整体式Co-Mo/Al2O3催化剂上的HDS的L-H型动力学方程。研究中综合考虑了反应物在催化剂上的吸附与脱附,回归计算出吸附平衡常数、吸附热、反应活化能和指前因子等参数。
直接脱硫反应(σ活性位):
(4)
间接脱硫反应(τ活性位):
(5)
Orozco等[46]在研究二苯并噻吩的HDS动力学时考虑了H2S抑制作用,并运用此模型解释了不同H2S分压对HDS的影响。以L-H方程为出发点,推导了二苯并噻吩在催化剂上加氢脱硫机理型反应反动学模型如下:
当PH2S>250 Pa时:
(6)
当PH2S<250 Pa时:
(7)
Orozco对此模型做以下假设:(1)反应过程中存在稳定的S2-和不饱和的金属Mo离子两种活性中心;(2)两种活性中心MD和S2-的活性中心数相等;(3)初始负电荷量等于S2-活性中心数目的两倍:(4)H2和H2S在MoS2上吸附后,与不饱和的金属Mo离子相互作用发生异裂解离,在反应过程中这种异裂解离受H2S浓度的影响;(5)若两种活性中心中M和S2-的任何一种吸附中心位吸附饱和后了,则H2、H2S的异裂终止。
4 结 语
近几年,随着煤焦油产量的不断增加和人们对环境问题的重视,煤焦油加氢改质化生产清洁油品具有重要意义。到目前为止,针对煤焦油加氢脱硫研究,众多学者主要研究含硫模型化合物的加氢脱硫反应机理及动力学,但并未对不同反应的差异进行对比和探究,且针对实际油品的研究比较少,以后可以从这方面加以研究和改进,以期为实际生产提供科学依据。
[1]方向晨,关明华,廖士纲.加氢精制[M].北京:中国石化出版社,2006:1-5.
[2]水恒福,张德祥,张超群.煤焦油分离与精制[M].北京:化学工业出版社,2007:9-10.
[3]高晋生,煤焦油及其产品《化工百科全书》编辑委员会,化学工业出版社《化工百科全书》编辑部. 化工百科全书:第11卷[M].北京:化学工业出版社,1996:45-47.
[4]肖瑞华.煤焦油化工学[M].北京:化学工业出版社,2007:98-100.
[5]薛新科.陈启文.煤焦油加工技术[M].北京:化学工业出版社,2007:23-35.
[6]Ted Oyamaa S, Yong-Kul Lee. The active site of nickel phosphide catalysts for the hydrodesulfurization of 4,6-DMDBT[J]. Journal of Catalysis, 2008, 258 (2):393-400.
[7]Kilanowski D R, Gates B C. Kinetics of hydrodesulfurization of benzothiophene catalyzed by sulfide Co-Mo/Al2O3[J]. Journal of Catalysis, 1980, 62(2): 70-78.
[8]Lipsch J M J G, Schuit G C A. The CoO-MoO3/γ-Al2O catalyst. III. Catalytic properties[J]. Journal of Catalysis, 1969, 15(3): 179-189.
[9]Kolboe S. Catalytic hydrodesulfurization of thiophene. VII. Comparison between thiophene, tetrahydrothiophene, and n-butanethiol[J]. Canadian Journal of Chemistry, 1969, 47(2): 352-355.
[10]Hensen E J M, Vissenberg M J, De Beer V H J, et al . Kinetics and mechanism of thiophenehydrodesulfurization over carbonsupported transition metal sulfides[J]. Journal of catalysis, 1996, 163(2): 429-435.
[11]Weigand B C, Friend C M. Model studies of the desulfurization reactions on metal surfaces and in organometallic complexes[J]. Chemical Reviews, 1992, 92(4): 491-504.
[12]Zaera F, Kollin E B, Gland J L. Vibrational characterization of thiophene decomposition on the Mo(100) surface[J]. Surface science, 1987, 184(1): 75-89.
[13]Liu A C, Friend C M. Evidence for facile and selective desulfurization: the reactions of 2,5-dihydrothiophene on Mo(110)[J]. Journal of the American Chemical Society, 1991, 113(3): 820 -826.
[14]Markel E J, Schrader G L, Sauer N N, et al. Thiophene, 2,3-and 2,5-dihydrothiophene, and tetrahydrothiophene hydrodesulfuri-zation on Mo and Re/γ-Al2O3catalysts[J]. Journal of Catalysis, 1989, 16(2): 11-22.
[15]Neurock M, van Santen R A. Atomic and molecular oxygen as chemical precursors in the oxidation of ammonia by copper[J]. Journal of the American Chemical Society, 1994, 116(15): 4427-4439.
[16]Angelici R J. Organometallic complexes as models for the adsorption of thiophenes on hydrodesulfurization(hds) catalysts[J]. Bulletin des Societes Chimiques Belges, 1995, 104(4-5): 265-282.
[17]Desikan P, Amberg C H. Catalytic hydrodesulphurization of thiophene: v. the hydrothiophenes selective poisoning and acidity of the catalyst surface[J]. Canadian Journal of Chemistry, 1964, 42(4): 843-850.
[18]Sullivan D L, Ekerdt J G. Mechanisms of thiophene hydrodesulfurization on model molybdenum catalysts[J]. Journal of Catalysis, 1998, 178(1): 226-233.
[19]Friend C M, Chen D A. Fundamental studies of hydrodesulfurization by metal surfaces[J]. Polyhedron, 1997, 16(18): 3165-3175.
[20]Moser W R, Rossetti J G A, Gleaves J T. Tetrahydrothiophene desulfurization on Co-Mo/γ-Al2O3: A temporal analysis of products(TAP) investigation[J]. Journal of Catalysis, 1991, 127(1): 190-200.
[21]Hensen E J M, Vissenberg M J, Debeer V H J, et al. Kinetics and mechanism of thiophene hydrodesulfurization over carbon supported transition metal sulfides[J]. Journal of Catalysis, 1996, 163(2): 429-435.
[22]Van Parijs I A, Froment G F. Kinetics of hydrodesulfurization on a Co-Mo/γ-Al2O3catalyst. I: Kinetics of the hydrogenolysis of thiophene[J]. Industrial & Engineering Chemistry Research, 1986, 25(2): 431-436.
[23]Gates B C, Sapre A V. Hydrogenation of aromatic compounds catalyzed by sulfided CoO-MoO3/γ-Al2O3[J]. Journal of Catalysis, 1982, 73(1): 45-49.
[24]Bartsch R, Tanielian C. Hydrodesulfurization: I. Hydrogenolysis of benzothiophene and dibenzothiophene over CoO-MoO3/γ-Al2O3catalyst[J]. Journal of Catalysis, 1974, 35(3): 353-359.
[25]Rollman L D. Catalytic hydrogenation of model nitrogen, sulfur, and oxygen compounds[J].Journal of Catalysis, 1977, 46(1):243-252.
[26]Houalla M, Broderick D H, Sapre A V, et al. Hydrodesulfurization of methyl-substituted dibenzothio-phenes catalyzed by sulfided Co-Mo/γ-Al2O3[J]. Journal of Catalysis, 1980, 61(2): 523 -527.
[27]李大东. 加氢处理工艺与工程[M]. 北京:中国石化出版社,2004:232-238.
[28]Satterfield C N, Roberts G W. Kinetics of thiophene hydrogenolysis on a cobalt molybdate catalyst[J]. American Institute of Chemical Engineers Journal, 1968, 14(1): 159-164.
[29]Kilanowski D R, et al. Kinetics of hydrodesulfurization of benzothiophene catalyzed by sulfided Co-Mo/Al2O3[J]. Journal of catalysis, 1980, 62(1): 70-78.
[30]Broderick D H, Gates B C. Hydrogenolysis and hydrogenation of dibenzothiophene catalyzed by sulfided CoO-MoO3/γ-Al2O3: The reaction kinetics[J]. American Institute of Chemical Engineers, 1981, 27(3): 663-673.
[31]Edvinsson, Irandoust S. Hydrodesulfurization of dibenzothiophene in a monolithic catalyst reactor[J]. Industrial & Engineering Chemistry Research, 1993, 32(2): 391-395.
[32]Orozco E O, Vrinat M. Kinetics of dibenzothiophene hydrodesulfurization over MoS2supported catalysts: modelization of the H2S partial pressure effect[J]. Applied Catalysis, 1998, 170(2): 195-206.
Research Progress on the Study of Carl Tar Hydrodesulfurization
HU Wei-yue1, LI Zhen2, CUI Wen-gang3, LI Dong3, LI Wen-hong3
(1 Shaanxi Province Supervision and Inspection Institute of Product Quality, Shaanxi Xi’an 710048;2ShaanxiPetrochemicalResearchandDesignInstitute,ShaanxiXi’an710054;3SchoolofChemicalEngineering,NorthwestUniversity,ShaanxiXi’an710069,China)
The type of sulfur-containing compounds in coal tar feedstock were reviewed, including thiols, sulfides, disulfides, thiophene, benzothiophene, dibenzothiophene and other sulfur compounds. The reaction mechanism of different types of sulfur compounds in the hydrodesulfurization process was summed up. In addition, the reaction kinetics of coal tar in a typical hydrodesulfurization of sulfur-containing compounds was analyzed. The research direction of hydrodesulfurization process was put forward. It aimed to provide theoretical guidance for the hydrodesulfurization of coal tar refining process, avoid the blindness of refined coal tar hydrogenation catalyst research and development.
hydrodesulfurization; sulfur compounds; reaction mechanism; kinetics
李稳宏(1955-),男,博士生导师。
TQ524
A
1001-9677(2016)016-0030-04
猜你喜欢
煤炭学报(2022年11期)2023-01-07
西南石油大学学报(自然科学版)(2021年3期)2021-07-16
环境保护与循环经济(2021年12期)2021-03-16
石油与天然气化工(2019年1期)2019-03-06
资源节约与环保(2017年10期)2017-01-22
山东工业技术(2016年16期)2016-08-15
中国资源综合利用(2016年7期)2016-02-03
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
橡胶工业(2015年2期)2015-07-29
能源(2014年9期)2014-09-15
