
木犀草素和木犀草苷在大鼠体内的药动学研究Δ
2016-09-23 01:20:47邹晓华王双虎周云芳
中国药房 2016年22期
邹晓华,王双虎,周云芳#
(1.丽水市第二人民医院药剂科,浙江丽水 323000;2.丽水市人民医院临床药学实验室,浙江丽水 323000)
木犀草素和木犀草苷在大鼠体内的药动学研究Δ
邹晓华1*,王双虎2,周云芳2#
(1.丽水市第二人民医院药剂科,浙江丽水323000;2.丽水市人民医院临床药学实验室,浙江丽水323000)
目的:建立测定木犀草素和木犀草苷在大鼠体内的药动学方法并测定其药动学参数。方法:16只SD大鼠随机分为木犀草素组(舌下iv,1.34 mg/kg)和木犀草苷组(舌下iv,0.64 mg/kg),并于给药前和给药后0、15、30 min及1、2、3、4、6、8、12、24、48 h经尾静脉取血0.5 ml制备血浆,采用超高效液相色谱串联三重四级杆质谱法测定血药浓度,并计算药动学参数。色谱柱为CORTECSTM UPLC®C1(8100 mm×2.1 mm,1.6 μm),流动相为乙腈-水(含0.1%甲酸),流速为0.4 ml/min,柱温为40℃,内标为槲皮苷。结果:木犀草素、木犀草苷的线性范围分别为2.5~500 ng/ml(r=0.998 2)、10~2 500 ng/ml(r=0.993 5),定量下限分别为1、2.5 ng/ml,提取回收率分别为70.75%~87.72%、75.40%~91.18%(n=6),日内、日间RSD均小于10%(n=3)。药动学参数t1/2分别为(1.88±0.32)、(1.57±0.08)h,CL分别为(0.77±0.18)、(0.06±0.01)L(/h·kg),AUC0-6 h分别为(189.60±40.04)、(1 093.14± 187.36)ng·h/ml,AUC0-∞分别为(195.18±38.37)、(1 097.11±188.07)ng·h/ml。结论:本研究建立的方法可用于木犀草素和木犀草苷在大鼠体内的药动学研究,两者在大鼠体内的药动学符合二室模型。
木犀草素;木犀草苷;血药浓度;药动学;超高效液相串联质谱法
木犀草素(Luteolin)是植物最常见的类黄酮类化合物并被归为黄酮类。含有木犀草素的植物如金银花、菊花、荆芥、白毛夏枯草等常作为食物或药物用于治疗各种疾病[1]。木犀草素的药理活性主要包括抗肿瘤、抗炎、抗感染、治疗糖尿病等[2-3]。目前,木犀草素的抗肿瘤活性已得到广泛研究,其还可有效抑制肝细胞核因子4α(HNF4α)而抑制其表达载脂蛋白B(apoB),但治疗糖尿病的机制尚不明确[4-6]。食用木犀草素可抑制肥胖、降低血清和肝脏脂质水平从而改善糖耐量高患者的饮食。木犀草苷(Cynaroside)是木犀草素-7-O-葡萄糖苷,是从许多药用植物中发现的黄酮类化合物,体外试验表明它能清除氧自由基、减少低密度脂蛋白的氧化[7]。早期研究报道显示木犀草苷在离体大鼠心脏中具有抗缺血作用,这可能与它的抗氧化性质有关。
木犀草素和木犀草苷血药浓度检测方法主要有高效液相色谱法[8-9]和高效液相串联质谱法[10-11]。本研究采用超高效液相色谱(UPLC)串联三重四级杆质谱法建立检测木犀草素和木犀草苷血药浓度的方法,研究了两者在大鼠体内的药动学过程。本研究建立的测定方法快速、稳定,可用于大鼠体内木犀草素和木犀草苷的药动学研究。
1 材料
1.1仪器
XEVO-TQD型UPLC-三重四极杆质谱联用仪(美国Wa-ters公司);Milli-Q A10型纯水系统(美国Millipore公司);5804R型低温高速离心机(德国Eppendorf公司);XS105DU型电子天平(美国梅特勒-托利多仪器有限公司);HH-6型数显恒温水浴锅(常州国华电器有限公司);TGL-16B型高速台式离心机(上海安亭科学仪器厂)。
1.2药品与试剂
木犀草素对照品、木犀草苷对照品、槲皮苷对照品(成都曼斯特公司,批号:MUST-15021005、MUST-15012204、MUST-14092105,纯度:99%);甲醇、乙腈(美国Merck Chemicals公司,色谱纯);其他试剂均为分析纯,均购自北京国药化学试剂公司。
1.3动物
清洁级SD大鼠16只,♀♂兼半,体质量(300±20)g,由温州医科大学实验动物中心提供,实验动物合格证号:SCXK(沪)2012-0002,实验动物使用许可证号:SYXK(浙)2010-0150。
2 方法与结果
2.1UPLC条件
色谱柱:CORTECSTM UPLC®C18(100 mm×2.1 mm,1.6 μm);流动相:乙腈-水(含0.1%甲酸),梯度洗脱(0~2 min 20%~95%乙腈,2~2.5 min 95%乙腈,2.5~2.6 min 95%~20%乙腈,2.6~3 min 20%乙腈);流速:0.4 ml/min;柱温:40℃;进样量:2 μl;内标:槲皮苷。
2.2质谱条件
电喷雾离子化源(ESI),负离子检测模式。毛细管电压:1.0 kV;木犀草素、木犀草苷、槲皮苷的锥孔电压:56、60、43 V,碰撞电压:30、26、20 V;源温度:150℃;脱溶剂气温度:500℃,锥孔气流量:50 L/h;脱溶剂气流量:1 000 L/h;氩气流量:0.15 ml/min。用于定量分析的离子质荷比:m/z 284.98→133.03(木犀草素)、m/z 447.05→284.96(木犀草苷)、m/z 301.12→151.05(槲皮苷)。
2.3溶液的制备
2.3.1对照品溶液的制备精密称取木犀草素和木犀草苷对照品各约5 mg,置于10 ml量瓶中,加入乙腈-水(1∶9,V/V)稀释至刻度,摇匀,得木犀草素和木犀草苷质量浓度均为500 μg/ml的对照品贮备液,4℃贮藏,备用。将木犀草素和木犀草苷对照品贮备液连续以纯水稀释成质量浓度为0.025、0.05、0.1、0.25、0.5、1、2.5、5 μg/ml的系列木犀草素对照品溶液和0.1、0.25、0.5、1、2.5、5、10、25 μg/ml的系列木犀草苷对照品溶液,4℃贮藏,备用。
2.3.2内标溶液的制备精密称取槲皮苷对照品10.01 mg,置于100 ml量瓶中,用甲醇溶解并稀释成质量浓度为100 μg/ml的内标贮备液。吸取1 ml内标贮备液置于100 ml量瓶中,加入甲醇稀释至刻度,摇匀,即得质量浓度为1 μg/ml内标溶液,4℃贮藏,备用。
2.3.3质控样品的制备分别吸取不同质量浓度的对照品溶液10 μl,置于1.5 ml塑料离心管中,加入空白血浆100 μl,涡旋30 s,制备得3、50、400 ng/ml的木犀草素和12、250、1 000 ng/ml的木犀草苷质控样品(即低、中、高浓度的质控样品)。
2.4血浆处理方法
取大鼠血浆100 μl,加入20 μl内标溶液,再加入200 μl冰乙腈沉淀并涡旋2 min,将样品于4℃下,以离心半径为6 cm、13 000 r/min离心10 min,取200 μl上清于进样瓶中,进样量为2 μl。
2.5系统适用性与专属性考查
分别取空白血浆、空白血浆+木犀草素和木犀草苷对照品+内标、给药1 h后大鼠血浆样品+内标,按“2.4”项下方法处理后,按“2.1”“2.2”项下条件进样测定,记录色谱图,见图1。结果表明,血浆内源性成分不干扰木犀草素和木犀草苷的测定,木犀草素的保留时间为1.61 min,木犀草苷的保留时间为1.04 min,内标的保留时间为1.63 min,3种物质在各自的离子通道中没有干扰。
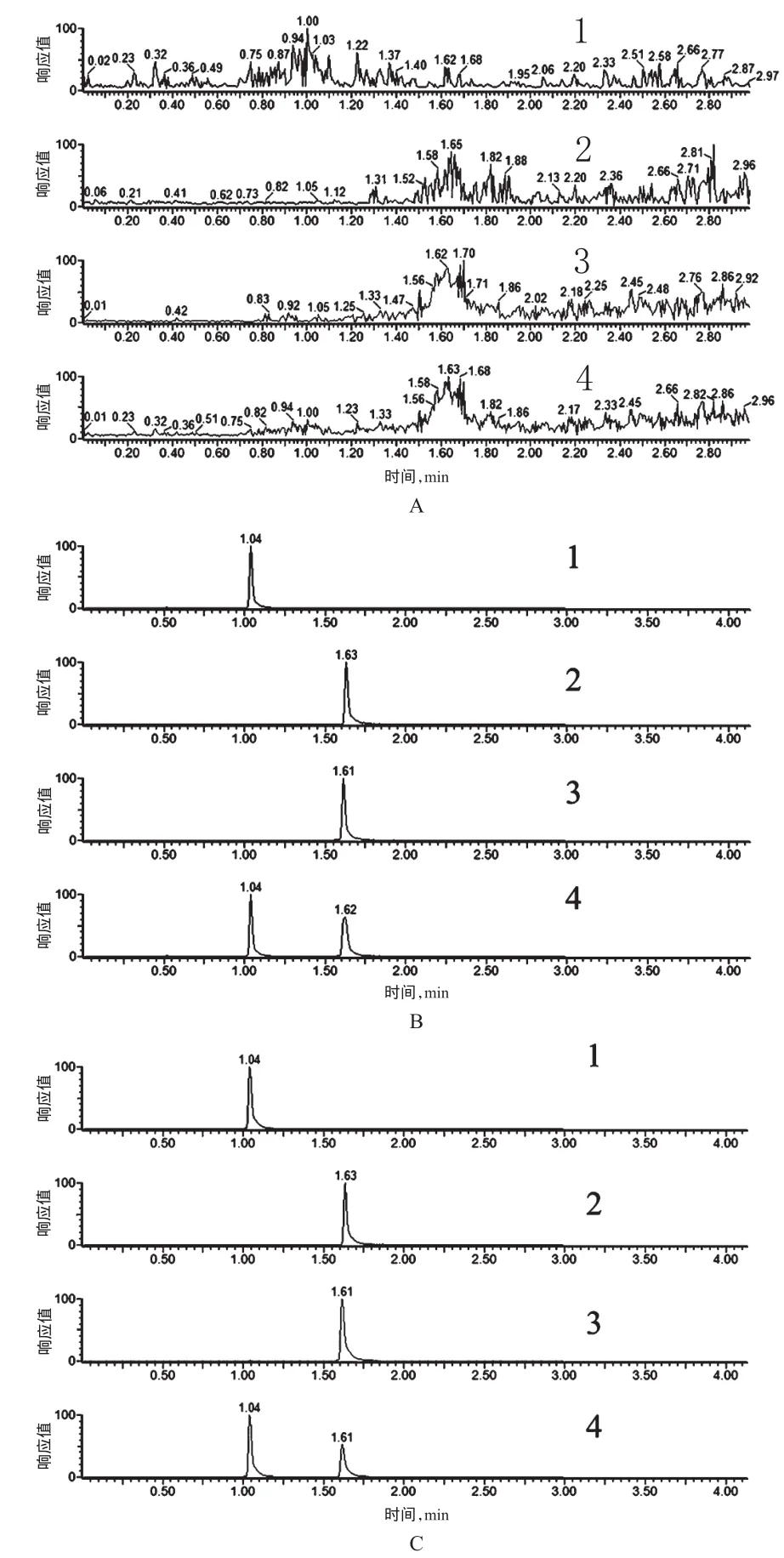
图1 UPLC图A.空白血浆;B.空白血浆+木犀草素和木犀草苷对照品+内标;C.给药1 h后大鼠血浆样品+内标;1.木犀草苷;2.槲皮苷;3.木犀草素;4.总离子流图Fig 1 UPLC chromatogramsA.blank plasma;B.blank plasma+reference substances of luteolin and cynaroside+internal standard;C.plasma sample collected at 1 h after administration+internal standard;1.cynaroside;2.quercetin;3.luteolin;4.TIC
2.6标准曲线的制备
按“2.3.1”项下方法制备对照品系列溶液,按“2.4”项下方法处理,按“2.1”“2.2”项下条件进样测定,记录色谱图。以木犀草素和木犀草苷的质量浓度(x,ng/ml)为横坐标,两者和内标峰面积的比值(y)为纵坐标,进行线性回归,得到木犀草素、木犀草苷的回归方程分别为y=6.545 93e-5x+0.001 331 02(r= 0.998 2)、y=3.086 54e-5x-0.004 089 43(r=0.993 5)。结果表明,木犀草素和木犀草苷质量浓度分别在2.5~500、10~2 500 ng/ml范围内和内标峰面积比值呈良好线性关系,定量下限分别为1、2.5 ng/ml(RSD分别为5.95%、7.43%)。
2.7精密度试验
按“2.3.3”项下方法制备低、中、高浓度的木犀草素和木犀草苷质控样品各6份,按“2.4”项下方法处理,按“2.1”“2.2”项下条件分别在同日内和连续3 d内进样测定。木犀草素和木犀草苷的日内、日间精密度试验结果见表1。
表1 精密度试验结果(±s,n=3)Tab 1 Results of precision test(±s,n=3)

表1 精密度试验结果(±s,n=3)Tab 1 Results of precision test(±s,n=3)
成分 加入量,ng/ml木犀草素3木犀草苷50 400 12 250 2 000日内精密度测得量,ng/ml 3.12±0.28 48.04±3.47 382.15±30.84 12.64±1.04 247.21±15.69 1 937.87±108.38 RSD,% 8.84 7.23 8.07 8.20 6.35 5.59日间精密度测得量,ng/ml 3.12±0.17 47.66±3.75 384.23±26.03 12.84±1.03 250.63±5.97 1 951.20±93.35 RSD,% 5.43 7.87 6.78 8.05 2.38 4.78
2.8方法回收率试验
按“2.3.3”项下方法制备低、中、高浓度的木犀草素和木犀草苷质控样品各6份,按“2.4”项下方法处理,按“2.1”“2.2”项下条件进样测定,以测得值与加入量的比值计算方法回收率。结果,木犀草素和木犀草苷低、中、高浓度血浆样品的方法回收率分别为104%、95.3%、96.06%、106.98%、100.25%、97.56%。方法回收率试验结果见表2。
表2 方法回收率试验、提取回收率试验和基质效应结果(±s,n=6)Tab 2 Results of recovery,extraction recovery and matrix effect test(±s,n=6)
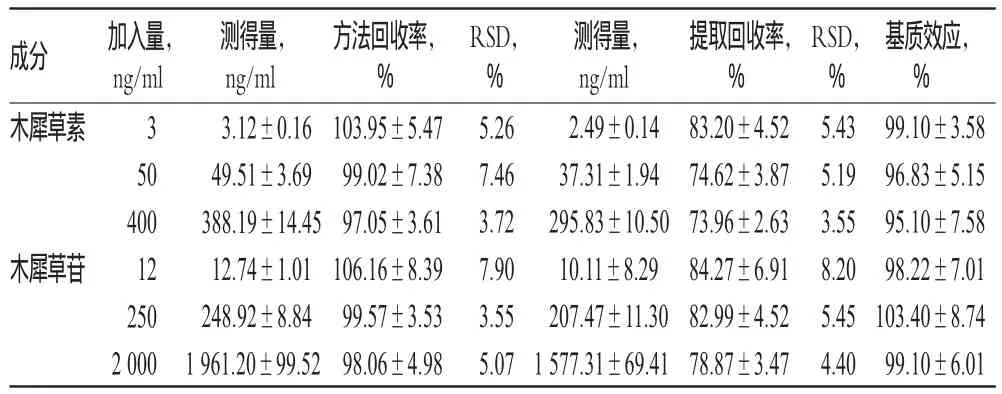
表2 方法回收率试验、提取回收率试验和基质效应结果(±s,n=6)Tab 2 Results of recovery,extraction recovery and matrix effect test(±s,n=6)
成分木犀草素木犀草苷加入量,ng/ml 3 50 400 12 250 2 000测得量,ng/ml 3.12±0.16 49.51±3.69 388.19±14.45 12.74±1.01 248.92±8.84 1 961.20±99.52方法回收率,% 103.95±5.47 99.02±7.38 97.05±3.61 106.16±8.39 99.57±3.53 98.06±4.98 RSD,% 5.26 7.46 3.72 7.90 3.55 5.07测得量,ng/ml 2.49±0.14 37.31±1.94 295.83±10.50 10.11±8.29 207.47±11.30 1 577.31±69.41提取回收率,% 83.20±4.52 74.62±3.87 73.96±2.63 84.27±6.91 82.99±4.52 78.87±3.47 RSD,% 5.43 5.19 3.55 8.20 5.45 4.40基质效应,% 99.10±3.58 96.83±5.15 95.10±7.58 98.22±7.01 103.40±8.74 99.10±6.01
2.9提取回收率和基质效应
按“2.3.3”项下方法制备低、中、高浓度的木犀草素和木犀草苷质控样品各6份,按“2.4”项下方法处理,按“2.1”“2.2”项下条件下进样测定,记录木犀草素和木犀草苷的峰面积A1。另取血浆样品100 μl,加入200 μl冰乙腈(含有10%的甲醇)沉淀并涡旋2 min,将样品在4℃下以离心半径为6 cm、13 000 r/min离心10 min;取上清液加入相应浓度的木犀草素和木犀草苷对照品溶液和内标溶液,配成低、中、高浓度的木犀草素和木犀草苷对照样品,不经任何处理,取2 μl进样检测,记录标准品的峰面积A2。计算血浆中木犀草素和木犀草苷的峰面积和不经任何处理的木犀草素和木犀草苷峰面积的比值,即得提取回收率,结果见表2。取不同浓度的木犀草素和木犀草苷对照品溶液,加入内标溶液,配成低、中、高浓度的木犀草素和木犀草苷对照品溶液(平行6个样品),取2 μl进样检测,记录对照品的峰面积A3,将已处理空白血浆配制的对照样品峰面积A2与相应浓度的对照品直接进样的峰面积A3比较,计算基质效应,结果见表2。由表2可知,基质基本不干扰检测,基质效应在85%~115%之间,均符合生物样品分析方法要求。
2.10稳定性试验
按“2.3.3”项下方法制备低、中、高浓度的木犀草素和木犀草苷质控样品各6份,按“2.4”项下方法处理,按“2.1”“2.2”项下条件分别于0、2、4、8、12、24 h取样测定,结果各浓度样品的RSD均小于5.0%,表明样品中木犀草素和木犀草苷在24 h内稳定。同法将3种浓度的质控样品置于-80℃冰箱中保存,分别于第1、5、15、30 d解冻,依法测定;经过3次反复冻融,每次间隔12 h,依法测定。结果各浓度样品的RSD均小于5.0%,表明样品在-80℃条件保存30 d内及反复冻融条件下稳定性较好。
2.11药动学研究
取SD大鼠16只随机分为木犀草素组(1.34 mg/kg)和木犀草苷组(0.64 mg/kg),单次舌下iv给药。于给药前和给药后0、15、30 min及1、2、3、4、6、8 h经大鼠尾静脉取血0.5 ml,置于含有肝素的EP管中,以离心半径为6 cm、3 000 r/min离心10 min,取血浆于-20℃冷冻保存。为保证大鼠血量充足,2 h后大鼠可以自由饮水。血浆按“2.4”项下方法处理后,按“2.1”“2.2”项下条件进样测定,记录色谱图。结果,木犀草素和木犀草苷在大鼠体内的浓度-药时曲线见图2。血药浓度数据采用DAS 3.0软件进行分析。结果显示木犀草素和木犀草苷在大鼠体内的消除过程属于一级消除,AUC0-6 h与剂量呈良好的线性关系,符合二室模型,详见表3。

图2 血药浓度-时间曲线Fig 2 Plasma concentration-time curves

表3 药动学参数Tab 3 Pharmacokinetic parameters
3 讨论
本实验中木犀草素和木犀草苷血药浓度检测采用的是UPLC-三重四极杆质谱联用仪,具有检测灵敏度高、检测时间短等优点,通常检测一个样本仅需要3 min,适合于高通量的药动学实验。文中涉及的方法学考察参考美国食品与药品监督管理局(FDA)[12]的方法和欧洲药品管理局(EMA)2012年2月1日开始实施的《生物样品分析方法验证指南》。本实验采用了沉淀蛋白的方法,先后用高氯酸、甲醇和乙腈沉淀蛋白,结果高氯酸酸性较强、对质谱影响较大[13-14],甲醇沉淀在质谱中容易出现溶剂峰而影响出峰时间,而乙腈沉淀则不会出现上述问题[15-16]。为了保证大鼠血流量充足,2 h后大鼠可以进行自由饮水,大大减少大鼠采血的压力,保证了每个采血时间点的准确性。流动相选择方面先后考察了甲醇和乙腈作为有机相,水、0.1%甲酸水、0.2%甲酸水、0.3%甲酸水以及0.5%的甲酸水作为水相,结果发现乙腈和0.1%甲酸水配制的流动相具有较好的分离度和响应值。选择槲皮苷作为内标是因为槲皮苷的结构与木犀草素和木犀草苷相似并且在样品处理过程中具有良好的回收率。检测方面,槲皮苷、木犀草素、木犀草苷都是负离子检测模式并且两两之间具有较好的分离度。
药动学参数显示,木犀草素和木犀草苷在大鼠体内的分布和消除半衰期较长,在体内的平均滞留时间较短;两者在大鼠体内呈现特征性的二室动力学特征;两者在大鼠体内的平均MRT分别为1.27、1.36 h,提示其在大鼠体内的滞留时间较短。木犀草素和木犀草苷均表现出了非线性药动学特征。
综上所述,该方法可快速、稳定地检测大鼠血浆中木犀草素和木犀草苷的浓度,为其临床合理应用提供依据。
[1]López-Lázaro M.Distribution and biological activities of the flavonoid luteolin[J].Mini Rev Med Chem,2009,9 (1):31.
[2]Li F,Wong TY,Lin SM,et al.Coadministrating luteolin minimizes the side effects of the aromatase inhibitor letrozole[J].J Pharmacol Exp Ther,2014,351(2):270.
[3]胡春萍,蔡雪婷,胡婷婷,等.木犀草素诱导非小细胞肺癌细胞株A549凋亡和G2周期阻滞[J].中国中药杂志,2012,37(9):1 259.
[4]Huang X,Dai S,Dai J,et al.Luteolin decreases invasiveness,deactivates STAT3 signaling,and reverses interleukin-6 induced epithelial-mesenchymal transition and matrix metal-loproteinase secretion of pancreatic cancer cells[J].Onco Targets Ther,2015,8:2 989.
[5]Kiselyuk A,Lee SH,Farber-Katz S,et al.HNF4α antagonists discovered by a high throughput screen for modulators of the human insulin promoter[J].Chem Biol,2012,19(7):806.
[6]Li LP,Wu XD,Chen ZJ,et al.Interspecies difference of luteolin and apigenin after oral administration of chrysanthemum morifolium extract and prediction of human pharmacokinetics[J].Pharmazie,2013,68(3):195.
[7]Sun X,Sun GB,Wang M,et al.Protective effects of cynaroside against H2O2-induced apoptosis in H9c2 cardiomyoblasts[J].J Cell Biochem,2011,112(8):2 019.
[8]陈林.RP-HPLC法测定独一味胶囊中木犀草苷的含量[J].中国药房,2011,22(36):3 447.
[9]姚文冰.HPLC法同时测定痰热清注射液中木犀草苷和黄芩苷的含量[J].中国药房,2011,22(35):3 311.
[10] Yin R,Han F,Tang Z,et al.Uflc-ms/ms method for simultaneous determination of luteolin-7-o-gentiobioside,luteolin-7-o-beta-d-glucoside and luteolin-7-o-beta-d-glucuronide in beagle dog plasma and its application to a pharmacokinetic study after administration of traditional chinese medicinal preparation:kudiezi injection[J].J Pharm Biomed Anal,2013,72(1):127.
[11] Huang Y,Zhang P,He F,et al.Simultaneous determination of four bioactive flavonoids from Polygonum orientale L.in dog plasma by UPLC-ESI-MS/MS and application of the technique to pharmacokinetic studies[J].J Chromatogr B Analyt Technol Biomed Life Sci,2014 (957):96.
[12] US Department of Health and Human Services,Food and Drug Administration,Center for Drug Evaluation and Research(CDER),et al.Guidance for industry on bioanalytical method validation[EB/OL].[2015-08-20].http://www. gmpru.com/validation/method/bioanalyticalmethod/4252 fnl.pdf.
[13]陈健苗,王双虎,周云芳,等.UPLC-MS/MS法检测人血浆中莫西沙星浓度[J].中国现代应用药学,2014,31 (12):1 503.
[14]邹晓华,王双虎,胡国新,等.UPLC-MS/MS快速测定大鼠血浆中的氯沙坦及其代谢产物[J].中国现代应用药学,2014,31(6):727.
[15]吴春美,王双虎,胡国新,等.超高效液相串联质谱法测定大鼠血浆中西酞普兰及其代谢产物的血药浓度[J].中国临床药理学杂志,2014,30(12):64.
[16]雷旭伟,王双虎,胡国新,等.超高效液相串联质谱法快速测定CYP2C9酶活性[J].中国药师,2014,17(11):1 804.
(编辑:刘明伟)
Pharmacokinetic Study on Luteolin and Cynaroside in Rats
ZOU Xiaohua1,WANG Shuanghu2,ZHOU Yunfang2
(1.Dept.of Pharmacy,Lishui Second People's Hospital,Zhejiang Lishui 323000,China;2.Laboratory of Clinical Pharmacy,Lishui People's Hospital,Zhejiang Lishui 323000,China)
OBJECTIVE:To establish the method for pharmacokinetic study of luteolin and cynaroside in rats and to determine pharmacokinetic parameters.METHODS:16 SD rats were randomly divided into luteolin group(sublingual iv,1.34 mg/kg)and cynaroside group(sublingual iv,0.64 mg/kg).0.5 ml blood were collected before administration and 0,15,30 min and 1,2,3,4,6,8,12,24,48 h after administration respectively to prepare plasma.UPLC-TQ-MS was adopted to determine plasma concentration,and pharmacokinetic parameters were calculated.A CORTECSTM UPLC®C18(100 mm×2.1 mm,1.6 μm)column was used with mobile phase consisted of acetonitrile-water(containing 0.1%formic acid)at a flow rate of 0.4 ml/min,the column temperature was set at 40℃,and quercetin was used as internal standard.RESULTS:The linear range of luteolin and cynaroside were 2.5-500 ng/ml(r=0.998 2)and 10-2 500 ng/ml(r=0.993 5).The lowest quantitation limits were 1 and 2.5 ng/ml,and extraction were 70.75%-87.72%and 75.40%-91.18%(n=6);RSD of inter-day and intra-day were all lower than 10%(n=3).Pharmacokinetic parameters as t1/2were(1.88±0.32)and(1.57±0.08)h;CL were(0.77±0.18)and(0.06±0.01)L/(h·kg);AUC0-6 hwere (189.60±40.04)and(1 093.14±187.36)ng·h/ml;AUC0-∞were(195.18±38.37)and(1 097.11±188.07)ng·h/ml.CONCLUSIONS:The method can be used for pharmacokinetic study of luteolin and cynaroside in rats,and the pharmacokinetics of them in rats are in line with two-compartment model.
Luteolin;Cynaroside;Plasma concentration;Pharmacokinetics;UPLC-ESI-MS/MS
R969.1
A
1001-0408(2016)22-3058-04
10.6039/j.issn.1001-0408.2016.22.11
浙江省中西医结合学会临床药学科研专项资金(No.2014LYK007)
*副主任药师。研究方向:医院药学。电话:0578-2317726。E-mail:zouxiaohua2015@126.com
#主任药师,硕士。研究方向:临床药理学。电话:0578-2780158。E-mail:zyf2808@126.com
2015-09-10
2016-06-16)
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2020年10期)2020-11-20 07:56:54
天然产物研究与开发(2018年2期)2018-04-04 02:01:12
中成药(2017年12期)2018-01-19 02:06:56
西江月(2017年4期)2017-11-22 07:24:09
长春中医药大学学报(2017年1期)2017-04-16 05:56:45
小雪花·成长指南(2016年10期)2016-11-01 06:02:27
分析测试学报(2015年5期)2016-01-13 06:18:41
中国野生植物资源(2014年1期)2014-03-29 05:45:02
中成药(2014年10期)2014-02-28 22:29:40
郑州大学学报(理学版)(2012年4期)2012-03-25 13:59:09
