
基于XcmⅠ的T载体及其在合成生物学中的应用
2016-09-19 00:42段仰凯梁飞燕谈晓明吕雪峰
生物工程学报 2016年7期
段仰凯,梁飞燕,谈晓明,吕雪峰
基于Ⅰ的T载体及其在合成生物学中的应用
段仰凯1,2,梁飞燕1,2,谈晓明2,吕雪峰2
1 中国科学院大学,北京 100049 2 中国科学院青岛生物能源与过程研究所 中国科学院生物燃料重点实验室,山东 青岛 266101
为了节约成本和提高实验效率,以商用pMD18-T载体为基础,构建获得了pFL-XS-T载体,其具有符合Biobrick标准的串联Ⅰ-Ⅰ-Ⅰ-Ⅰ克隆序列,通过对Ⅰ的酶切处理即可以通过TA克隆方便地克隆PCR片段。克隆验证实验结果表明,经Ⅰ处理的该载体可以与PCR片段进行TA连接,连接效率及阳性率均可以满足实验室要求并且可以得到Biobrick标准质粒。此外,该载体不仅可以与其余Biobrick标准元件进行串联,还可以作为功能DNA元件的筛选载体。
Biobrick标准,TA克隆,合成生物学
传统的分子生物学实验中,由于受可用酶切位点的限制,许多重组后的DNA无法继续拼装,因而不能满足合成生物学的大量、快速拼装的要求。Knight发明了一套新的DNA组装策略并将其命名为“Biobrick”。该策略利用限制性内切酶的同尾酶,酶切后可以得到互补的粘性末端并且连接后形成的碱基序列无法被其中任何一种酶识别的原理,实现了DNA片段的循环组装[1-3]。经典的Biobrick拼接方法,需要先通过PCR克隆的方式,将目的片段克隆到两翼带有RⅠ-Ⅰ、Ⅰ-Ⅰ等酶切位点的质粒上。这就需要在扩增目的片段的引物末端加上Ⅰ和Ⅰ等酶切位点,引物上额外的酶切位点的添加使得合成成本升高且步骤相对繁琐。
PCR是分子生物学中获得体外DNA片段的最常用的方法,用DNA聚合酶PCR扩增得到片段的3′端突出一个A碱基[4],TA克隆是DNA聚合酶扩增得到的PCR产物与载体连接最快速最有效的方法之一[5-8]。商用T载体,如pMD18-T,载体两端有Ⅰ等多个酶切位点可用,然而通常条件下克隆得到的质粒并不具有Biobrick特征,在载体的一端缺少Ⅰ的同尾酶Ⅰ。若要利用TA克隆的方式获得Biobrick特征质粒,需要在目的片段的一端加入Ⅰ酶切位点且片段插入后需确保pMD18-T载体引入的Ⅰ和目的片段引入的Ⅰ酶切位点位于插入片段的两端。该方法虽然克隆效率高,但扩增片段的引物末端还需加入Ⅰ酶切位点且克隆后需要鉴定插入方向,这使得引物合成成本升高且步骤仍然相对繁琐。
由此可见,获得一个带有Biobrick酶切位点的T载体就可以解决传统Biobrick组件获取方法所面临的问题。许多研究报道过利用限制性内切酶可以构建T载体[9-11],其中Ⅰ构建T载体的研究尤其得到了广泛关注[12-16]。Ⅰ为一种Ⅱ型限制性内切酶,识别位点为5′-CCANNNNN | NNNNNTGG-3′,如果设计第5个N为T,Ⅰ酶切后在3′端突出一个T,可以与聚合酶反应产物上的3′端单A突出配对,直接进行亚克隆或测序。但是目前还没有利用Ⅰ构建符合Biobrick规范T载体的研究报道。事实上,pMD18-T与符合Biobrick规范T载体的主要区别只是在pMD18-T的d Ⅲ一端缺少Ⅰ酶切位点。因此,将一段一端带有Ⅰ酶切位点,另一端带有Ⅰ和Ⅰ酶切位点的PCR产物与pMD18-T载体经过TA克隆连接,会得到一个符合Biobrick规则的T载体质粒,得到的质粒再经过Ⅰ酶切便可以得到符合Biobrick规范的T载体 (图1),该载体不仅可以与DNA片段进行TA克隆连接,而且克隆后的片段还可以利用Biobrick规则进行后续组装 (图1)。因此,本文按照该方法构建了Biobrick-T载体并对该载体的克隆效率及应用的有效性进行了验证。
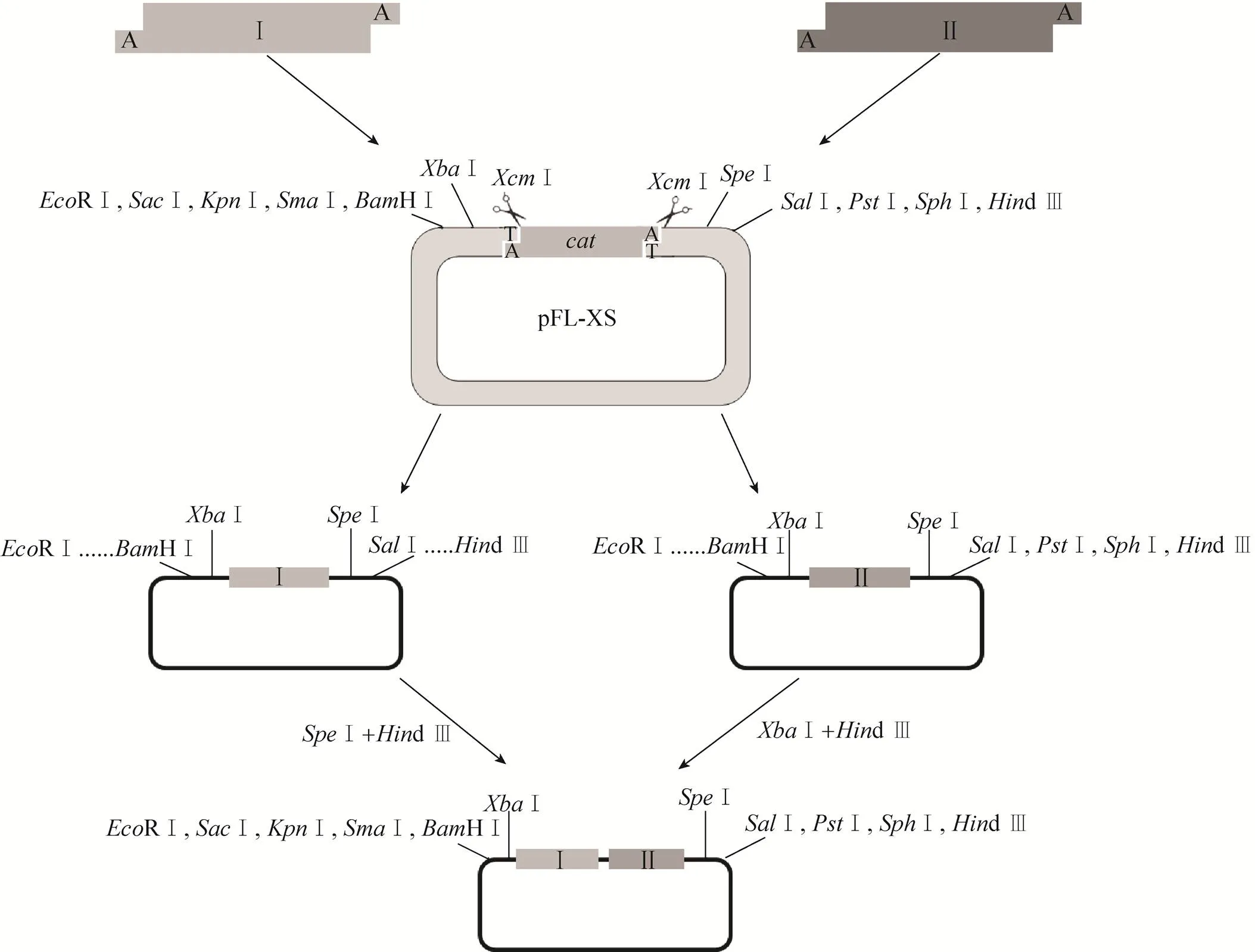
图1 基于XcmⅠ的T载体构建及其在合成生物学中应用示意图
1 材料与方法
1.1 试剂
pMD18-T载体和感受态细胞购于TaKaRa (日本)。其余用于分子克隆的试剂盒购于Omega (美国)。内切酶Ⅰ购于NEB (美国),其余所用的限制性内切酶和分子克隆所用到的相关的酶购于Thermo (加拿大)。
1.2 菌株和质粒的构建
本研究所用的引物见表1,用到的质粒见表2。
表1 本研究中所用的引物
Table 1 Primers used in this study

Underlined nucleotides represent the restriction enzyme sites
表2 本研究中所用的质粒
Table 2 Plasmids used in this study

1.2.1 使用pMD18-T载体构建Biobrick标准质粒
以pRL271[17]为模板,Cm-F (含Ⅱ酶切位点)/Cm-R (含Ⅰ酶切位点) 为引物PCR扩增得到约1 kb的氯霉素抗性基因片段,随后将该片段连接到pMD18-T载体中得到质粒pXT170c,形成Ⅰ和Ⅰ同尾酶的Biobrick标准的质粒。随后用Ⅰ酶切pRL57[18],得到的带有壮观霉素抗性编码基因的Omega片段与用Ⅱ和Ⅰ酶切并补平的pXT170c片段连接,得到有Biobrick标准的质粒pXT170s。用QucikChange方法消除壮观霉素抗性片段中的Ⅰ位点 (用引物Omega-qc-F/Omega-qc-R进行突变)得到质粒pXT170′。
1.2.2 Biobrick-T载体pFL-XS的构建
以pRL271[17]为模板,X-F1/X-R1为引物PCR扩增得到约1 kb的氯霉素抗性片段,随后将该片段连接到pMD18-T载体中得到质粒pXcm-XS。用Ⅰ酶切该质粒并用T4 DNA聚合酶补平,自连接,最终得到消除Ⅰ的质粒pFL-XS。
1.2.3 利用Biobrick规则进行质粒构建
如图2所示,使用Ⅰ和Ⅰ酶切质粒BBa_E0240得到绿色荧光报告基因片段 (eGFP),使用Ⅰ和Ⅰ酶切pFL-XS得到载体片段,将两个片段分别回收后连接得到质粒pXT238。用Ⅰ和Ⅰ酶切pXT238得到的载体与用Ⅰ和Ⅰ酶切pXT170′得到的壮观霉素抗性片段连接得到质粒pXT244′。
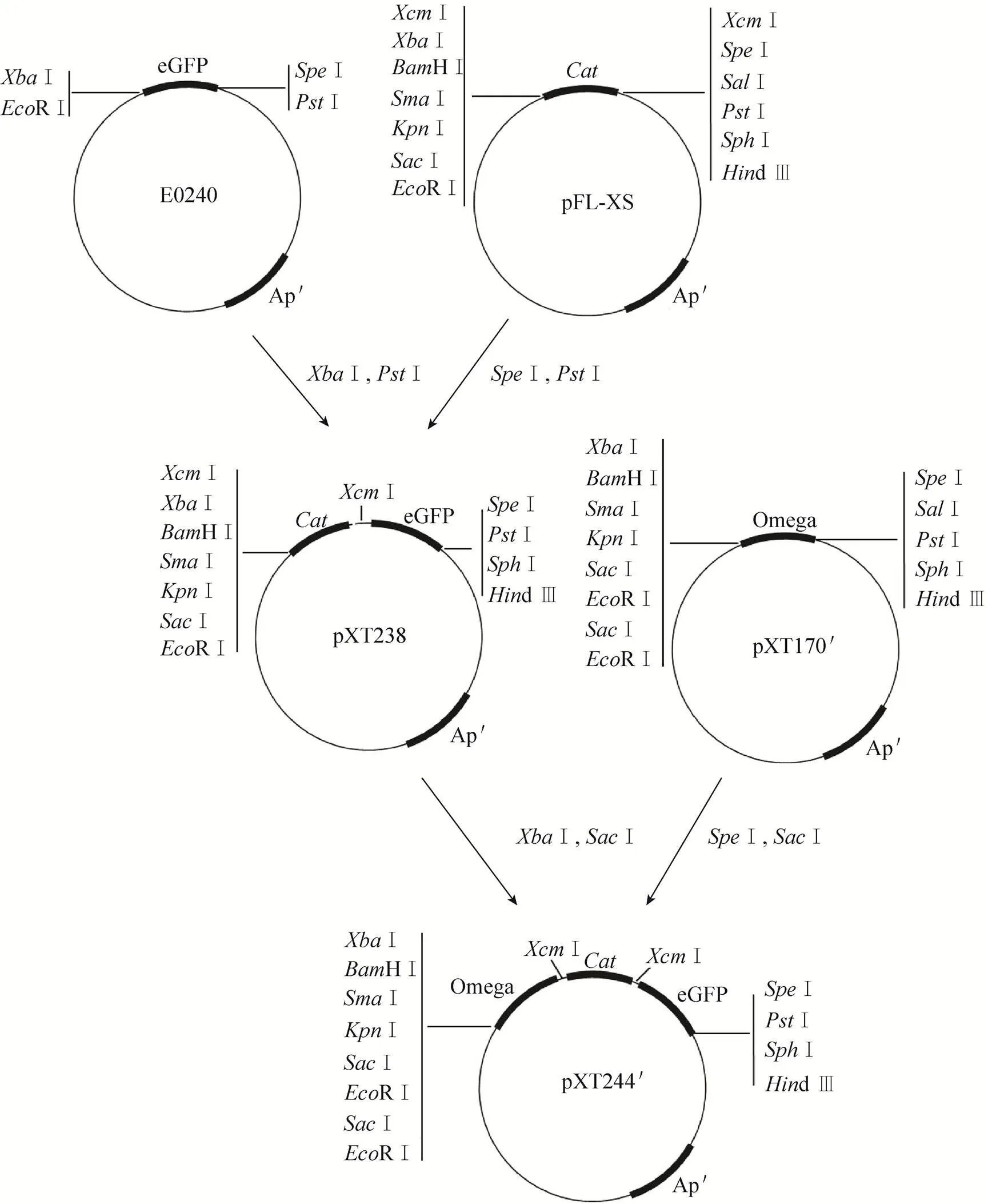
图2 利用Biobrick规则构建质粒pXT244′流程图
1.2.4 用于大肠杆菌中检测启动子活性的质粒构建
用Ⅰ酶切pXT244′得到有3′-T突出的2.7 kb的DNA片段,该片段可作为T载体。lacI-R/PlacO-R为引物,质粒pRL59EH[19]为模板用DNA聚合酶PCR得到3′端带有碱基A的lacI-Ptac片段,lacI-R/PlacO-R为引物,质粒pXT181b为模板用DNA聚合酶PCR得到3′端带有碱基A的lacI-Ptrc片段,将两个启动子片段分别与pXT244′得到的载体进行TA克隆分别得到含有Ptac启动子的质粒pFL75和含有Ptrc启动子的质粒pFL76。
1.3 pFL-XS-T载体克隆效率及阳性率测定
pFL-XS质粒用Ⅰ (NEB) 酶切并使用胶回收试剂盒 (OMEGA) 回收得到2.7 kb的pFL-XS-T载体 (40 μg/μL),实验中对照载体使用的是商业载体pMD18-T (TaKaRa,50 μg/μL)。
首先,实验使用插入片段 (pMD18-T试剂盒内置对照片段,50 μg/μL) 与两个载体分别进行连接。连接过夜,转化到大肠杆菌DH5α,分别随机挑取20个单克隆,使用通用引物RV-M和M13-47对20个单克隆进行菌落PCR鉴定阳性克隆。阳性克隆的标准是菌落PCR可以得到约660 bp的PCR产物。
其次,为了验证pFL-XS-T对随机克隆的PCR产物的连接效率,PCR扩增来源于集胞藻PCC 6803 (sp. PCC 6803) 328 bp和来源于聚球藻PCC 7942(sp. PCC 7942) 2 199 bp两段DNA片段。用胶回收试剂盒(OMEGA)回收,分别与pFL-XS-T载体连接过夜随后转化到大肠杆菌DH5α。分别随机挑取24个单克隆,用上述通用引物对单克隆进行菌落PCR。检验出阳性克隆的标准是菌落PCR可以分别得到约500 bp和2 350 bp的PCR产物。
1.4 大肠杆菌转化
将质粒pFL75、pFL76和pFL77转化大肠杆菌DH5α中,分别得到重组大肠杆菌FL178、FL179和FL180。
1.5 荧光显微镜观察
将重组大肠杆菌FL178、FL179和FL180接种到LB培养基中,在37 ℃培养至指数生长期时加入1 mmol/L的异丙基硫代半乳糖苷 (IPTG)。IPTG诱导后5 h,对不同菌的培养液取样并进行荧光显微镜 (Olympus BX51, 日本) 观察[20]。
2 结果与分析
2.1 T尾Biobrick载体的获得
构建得到pFL-XS质粒经过内切酶Ⅰ可以得到Biobrick-T载体。由图3可以清楚地看出,内切酶Ⅰ可以有效地将pFL-XS质粒酶切成一个2 722 bp的载体片段和一个962 bp的氯霉素抗性片段,并且两个片段的大小可以确保在回收胶上得到有效分离 (图3),其中2 722 bp片段为Biobrick-T载体片段。该载体以pMD18-T载体为基本框架,不仅具备各种多克隆位点,而且在载体两端分别带有RⅠ-Ⅰ和Ⅰ-Ⅰ Biobrick酶切位点,使得与该载体进行TA克隆后的DNA片段获得Biobrick特征(图1)。

图3 pFL-XS-T载体的XcmⅠ限制性内切酶酶切检测 结果
2.2 pFL-XS-T载体克隆效率及阳性率
商业载体pMD18-T转化平板上的单克隆数量 (大于200个) 远高于pFL-XS-T载体转化平板上的单克隆数量 (32个),但是自制pFL-XS-T浓度略低于商用载体浓度 (40 µg/mL VS 50 µg/mL),如果提高pFL-XS-T载体的浓度,转化后的单克隆数目有再增多的可能。但是在该浓度下,pFL-XS-T为载体的转化平板上的单克隆数目足够应用于实验室条件下目的克隆的获得。
本研究以菌落PCR检测转化子插入片段的方法来验证阳性克隆并计算阳性率,分别选用pMD18-T载体的商用插入片段和蓝藻基因组随机片段作为待测片段。在插入商用片段的实验中,随机挑取20个单克隆PCR鉴定,pMD18-T载体重组质粒的阳性率为100% (图4A),即所有单克隆中都成功得到了含有插入商业片段的重组质粒;pFL-XS-T载体重组质粒的阳性率为90% (20个单克隆中18个为阳性),即有10%的单克隆中没有成功得到含有插入商业片段的重组质粒 (图4B)。假阳性的出现是因为pFL-XS质粒在DNA琼脂胶上条带的位置与pFL-XS-T线性载体的条带基本重合 (数据未列出),与之前文献报道类似[21-22],受Ⅰ酶切效率的限制,有少量的没有受到Ⅰ酶切的质粒混入了载体片段中,该质粒含有氯霉素和氨苄青霉素的抗性,因此转化得到的克隆可以在氨苄青霉素的抗性板上生长,因此推测影响重组阳性率的主要因素是pFL-XS质粒的酶切完全程度。这个问题可以通过增加填充片段(本研究为氯霉素)的长度,使得载体片段和未被酶切的质粒在电泳胶上易于分开回收以及引入蓝白斑筛选来提高连接效率[21,23-25]。
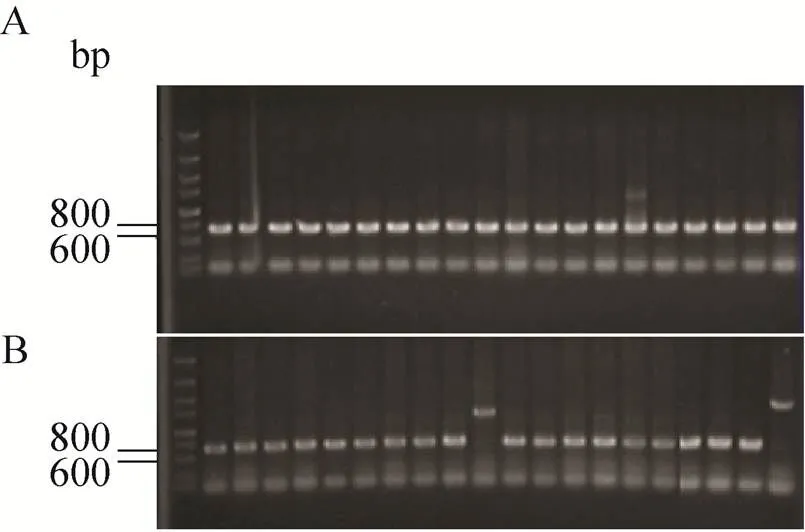
图4 商用插入片段与pFL-XS-T载体的TA克隆效率及阳性率检测
T载体的广泛应用是因为能够直接与实验室条件下用DNA聚合酶PCR得到的产物连接并进行后续的亚克隆和测序工作。为了验证pFL-XS-T对随机克隆的PCR产物的连接效率,用DNA聚合酶分别PCR克隆了来源于蓝藻基因组的328 bp和2.2 kb的基因片段,纯化后直接与pFL-XS-T载体连接过夜,转化。各随机挑取24个单克隆进行菌落PCR鉴定,发现两者的阳性率都高于75% (图5)。

图5 随机插入片段与pFL-XS-T载体的TA克隆效率及阳性率检测
以上结果说明该载体可以有效地通过TA克隆的方式连接DNA片段,这些片段连接以后形成的质粒即为Biobrick标准的质粒,这样便可以省去PCR扩增中为了加入Biobrick所需要的酶切位点而增加的引物长度。因此通过该方法可以很便捷的建立Biobrick元件库。
2.3 不同方式获得的符合Biobrick标准的基因元件的组装和应用验证
为了验证得到的pFL-XS在实际应用中是否可以与符合Biobrick标准的元件有效拼接。我们先将质粒pFL-XS与Biobrick标准质粒库中的质粒BBa_E0240进行Biobrick规则组装 (详见材料与方法1.2.3),得到了含有报告基因的质粒pXT238,该质粒仍然具有Biobrick特征。随后我们又将pXT238与之前构建的具有Biobrick特征的质粒pXT170′再次进行了Biobrick规则组装,最终得到了带有报告基因且仍然具有Biobrick特征的质粒pXT244′。质粒pXT244′构建过程不仅证明了本研究得到的pFL-XS载体质粒不仅具有Biobrick特征并且可以与其余Biobrick规则的片段及质粒进行连续的Biobrick规则组装。
另外,之前有研究将T载体用于高通量筛选[26],由于本研究得到的pFL-XS-T载体也具有T载体的特点,前面我们已经验证了该载体连接的有效性,为了进一步验证该载体应用的有效性,我们将该载体可进行TA克隆的特性用于启动子筛选。质粒pXT244′源于质粒pFL-XS,是以pFL-XS为载体,利用Biobrick规则与其余片段组装获得,该质粒经过Ⅰ酶切后可以得到含有报告基因的T载体,因此需要筛选的启动子片段只需要经过PCR和TA克隆便可以得到启动子筛选质粒。我们分别构建了含有Ptrc和Ptac启动子的筛选质粒及对照质粒 (见材料与方法) 并将得到的质粒转化到大肠杆菌中。培养的重组大肠杆菌在加入IPTG诱导以后,在日光中就可以看到含有Ptrc和Ptac启动子的菌株有荧光出现 (图6C)。在荧光显微镜下观察,很明显可以看出含有Ptrc和Ptac启动子的菌株可以发出绿色荧光 (图6A和图6B)。
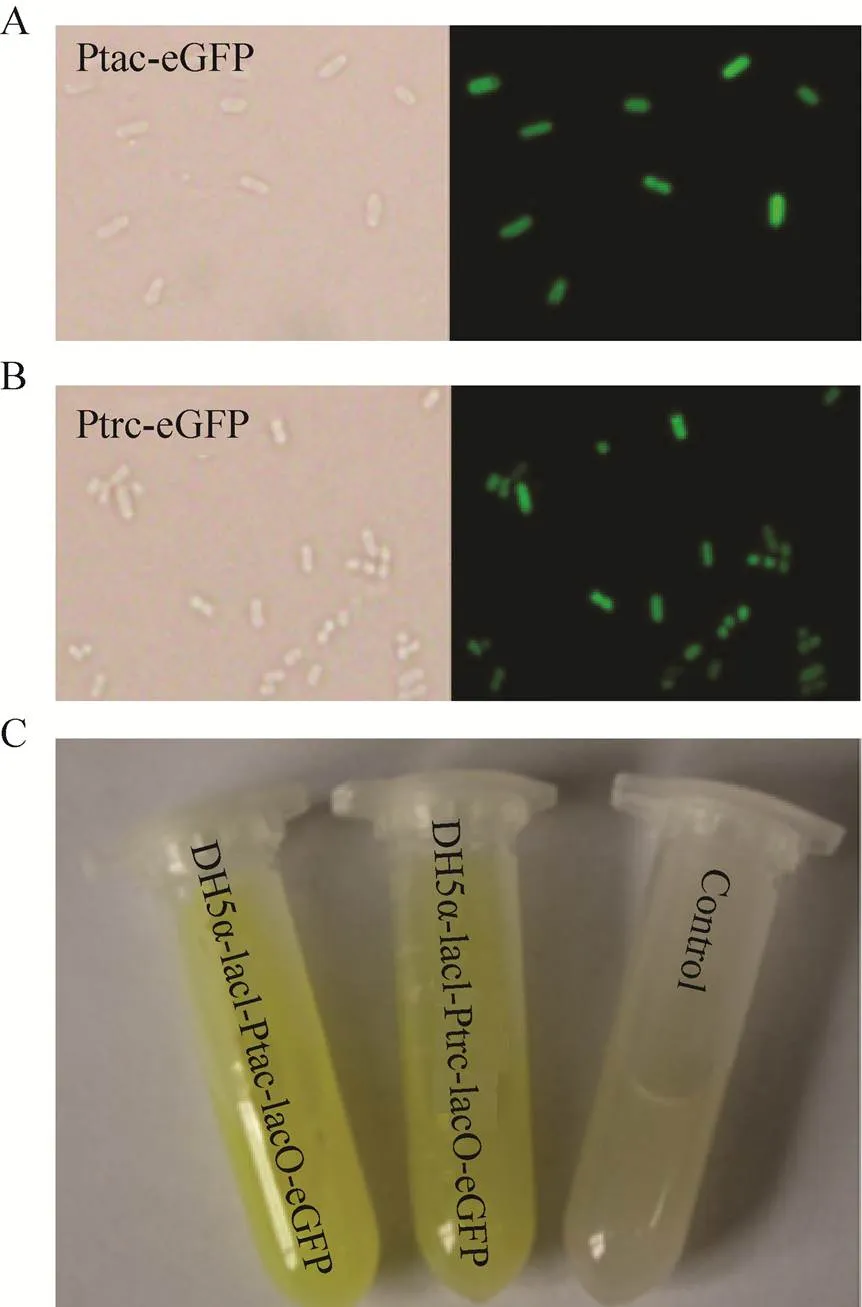
图6 重组大肠杆菌FL178、FL179和FL180的表型分析
因此,基于pFL-XS-T载体构建的质粒不仅可以实现与其他来源的Biobrick元件进行拼装,而且还可以作为T载体,通过TA克隆的方式对其中的元件进行建库及高通量筛选。
3 结果与讨论
标准化生物学元件可以大大简化生物基因工程过程。本研究通过利用Ⅰ对商业化载体pMD18-T的改造获得了具有Biobrick标准的pFL-XS-T载体,经实验验证,该载体与片段的连接及重组效率均可以满足实验需求。另外本研究获得的载体同时具备了Biobrick标准元件和T载体的功能,不仅可以与其他的Biobrick标准元件进行装配还可以作为T载体用于功能DNA元件的筛选,简化了传统Biobrick方法,减少了引物合成成本,为以后合成生物学研究提供了一种可选择的平台工具。
REFERENCES
[1] Sleight SC, Bartley BA, Lieviant JA, et al. In-Fusion BioBrick assembly and re-engineering. Nucleic Acids Res, 2010, 38(8): 2624–2636.
[2] Knight T. Idempotent Vector Design for Standard Assembly of Biobricks. Dspace @ MIT, 2003.
[3] Lee TS, Krupa RA, Hajimorad M, et al. Biobrick vectors and datasheets: a synthetic biology platform for metabolic engineering. Abstracts of Papers of the American Chemical Society, 2010, 239.
[4] Clark JM. Novel non-templated nucleotide addition reactions catalyzed by procaryotic and eucaryotic DNA-polymerases. Nucleic Acids Res, 1988, 16(20): 9677–9686.
[5] Marchuk D, Drumm M, Saulino A, et al. Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res, 1991, 19(5): 1154.
[6] Ichihara Y, Kurosawa Y. Construction of new T-vectors for direct cloning of PCR products. Gene, 1993, 130(1): 153–154.
[7] Holton TA, Graham MW. A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors. Nucleic Acids Res, 1991, 19(5): 1156.
[8] Mead DA, Pey NK, Herrnstadt C, et al. A universal method for the direct cloning of PCR amplified nucleic acid. Biotechnology, 1991, 9(7): 657–663.
[9] Wang BL, Liang H, Liu R, et al. Construction of a restriction-endonuclease-1105Ⅰ-generated T-vector for high-throughput cloning and expression. Biotechnol Appl Bioc, 2007, 48(1): 29–33.
[10] Zhao YF, Liu ZC, Yu SY, et al. Construction of a high efficiency PCR products cloning T vector using pGEM-5zf (+). Avicenna J Med Biotechnol, 2009, 1(1): 37–39.
[11] Dimov SG. pSDTV vector: a modification of the pBluescript SK + plus plasmid in order to perform PCR-fragments TA-cloning using1105Ⅰ restriction endonuclease. Mol Biol Rep, 2012, 39(5): 6133–6139.
[12] Harrison J, Molloy PL, Clark SJ. Direct cloning of polymerase chain reaction products in anⅠT-vector. Anal Biochem, 1994, 216(1): 235–236.
[13] Borovkov AY, Rivkin MI.I-containing vector for direct cloning of PCR products. Biotechniques, 1997, 22(5): 812–814.
[14] De Vries E. pUCPCR1-A vector for direct cloning of PCR products in a doubleI restriction site offering compatible single 3´-overhanging T residues. Mol Biotechnol, 1998, 10(3): 273–274.
[15] Arashi-Heese N, Miwa M, Shibata H.I site-containing vector for direct cloning andtranscription of PCR product. Mol Biotechnol, 1999, 12(3): 281–283.
[16] Park HK, Zeng CY. Construction of anⅠ-generated T vector bearing green fluorescent protein marker for direct cloning of PCR products. Anal Biochem, 2007, 360(1): 144–145.
[17] Black TA, Cai YP, Wolk CP. Spatial expression and autoregulation of, a gene involved in the control of heterocyst development in. Mol Microbiol, 1993, 9(1): 77–84.
[18] Elhai J, Wolk CP. A versatile class of positive-selection vectors based on the nonviability of palindrome-containing plasmids that allows cloning into long polylinkers. Gene, 1988, 68(1): 119–138.
[19] Fürste JP, Pansegrau W, Frank R, et al. Molecular cloning of the plasmid Rp4 primase region in a multi-host-rangeexpression vector. Gene, 1986, 48(1): 119–131.
[20] Wu W, Zhang L, Yao L, et al. Genetically assembled fluorescent biosensor fordetection of bio-synthesized alkanes. Scientific Reports 10907 2015.
[21] Jo C, Jo SA. A simple method to construct T-vectors usingⅠcassettes amplified by nonspecific PCR. Plasmid, 2001, 45(1): 37–40.
[22] Jun SY, Yoon SJ, Kang SH. One-step preparation of a TA-cloning vector from a specially designed parent plasmid containing a dualgene system. Mol Biotechnol, 2010, 45(1): 9–14.
[23] Gu JS, Ye CJ. pYEMF, a pUC18-derivedI T-vector for efficient cloning of PCR products. Mol Biotechnol, 2011, 47(3): 229–233.
[24] Janner CR, Brito ALP, Moraes LMP, et al. pPCV, a versatile vector for cloning PCR products. Springerplus, 2013, 2(1): 441.
[25] Hong SG, Choi JY, Pryor BM, et al. An efficient method to prepare PCR cloning vectors. Mycobiology, 2009, 37(3): 240–242.
[26] Berlec A, Štrukelj B. Generating a custom TA-cloning expression plasmid for. Biotechniques, 2012, 52(1): 51–53.
AnⅠ-generated T vector and its applications in synthetic biology
Yangkai Duan1,2, Feiyan Liang1,2, Xiaoming Tan2, and Xuefeng Lü2
1 University of Chinese Academy of Sciences, Beijing 100049, China 2 Key Laboratory of Biofuels, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, Shandong, China
For more economical and efficient DNA clonging, pFL-XS-T, a Biobrick-T vector was constructed based on pMD18-T vector, carrying clonging regions ofⅠ-Ⅰ-Ⅰ-Ⅰ. The results revealed that PCR products could be conveniently inserted into pFL-XS-T vevtor digested byⅠby means of TA cloning. The positive frequency of recombination can meet the experimental requirements and all the plasmids obtained meet Biobrick standard. Moreover, the pFL-XS-T is compatible with other Biobrick parts, and serves as a vector for functional DNA fragments screening.
Biobrick standard, TA cloning, synthetic biology
October 23, 2015; Accepted: November 16, 2015
生物技术与方法
段仰凯, 梁飞燕, 谈晓明, 等. 基于Ⅰ的T载体及其在合成生物学中的应用. 生物工程学报, 2016, 32(7): 956–965.
Duan YK, Liang FY, Tan XM, et al. AnⅠ-generated T vector and its applications in synthetic biology. Chin J Biotech, 2016, 32(7): 956–965.
Supported by: Natural Science Foundation of Shandong Province (No. ZR2013CQ045).
Corresponding authors: Xuefeng Lü. Tel: +86-532-80662629; Fax: +86-532-80662712; E-mail: lvxf@qibebt.ac.cn
山东省自然科学基金 (No. ZR2013CQ045) 资助。
(本文责编 郝丽芳)
猜你喜欢
中国特种设备安全(2022年1期)2022-04-26
空间科学学报(2021年1期)2021-05-22
中国现代医药杂志(2020年10期)2020-12-14
环境保护与循环经济(2017年5期)2018-01-22
中国核电(2017年2期)2017-08-11
现代检验医学杂志(2016年3期)2016-11-15
现代工业经济和信息化(2016年2期)2016-05-17
中国果菜(2016年9期)2016-03-01
中国蔬菜(2015年9期)2015-12-21
医学研究杂志(2015年3期)2015-06-10
