
HPLC同时检测酸性橙Ⅱ和碱性嫩黄O色谱条件优化
2016-09-18 12:38:37雷智棋张志清四川农业大学食品学院四川雅安6504重庆市涪陵食品药品检验所重庆408000
中国酿造 2016年1期
李 莎,雷智棋,朱 红,张志清(.四川农业大学 食品学院,四川 雅安 6504;.重庆市涪陵食品药品检验所,重庆 408000)
HPLC同时检测酸性橙Ⅱ和碱性嫩黄O色谱条件优化
李莎1,2,雷智棋2,朱红2,张志清1*
(1.四川农业大学 食品学院,四川 雅安 625014;2.重庆市涪陵食品药品检验所,重庆 408000)
该研究建立高效液相色谱法同时检测食品中酸性橙Ⅱ和碱性嫩黄O的方法,对高效液相色谱中色谱柱、柱温、流动相种类及比例、流速、波长等影响因素进行了研究及优化,得到色谱检测条件为:色谱柱为Kromasil C18(4.6mm×150mm,5μm);流动相为甲醇-50mmol/L乙酸铵溶液=50∶50(V/V);流速:1.0mL/min;检测波长450 nm;柱温35℃。在此最佳条件下,碱性嫩黄O及酸性橙Ⅱ的分离度好,且峰型正常。结果表明,该方法适合食品中碱性嫩黄O及酸性橙Ⅱ的检测。
食品;酸性橙Ⅱ;碱性嫩黄O;高液相色谱法
食用色素是以食品着色、改善食品的色泽为目的的食品添加剂,因其能赋予食品美好的外观而得以广泛添加[1]。酸性橙Ⅱ作为常用的酸性偶氮染料,早在20世纪初,科学家就发现用偶氮类化合物猩红色素喂养动物的肝癌发病率是100%,对人体危害极大。AM INK A等[2]研究发现,偶氮类染料不管是高剂量还是低剂量都会对人体产生不利的影响,并改变人体生化指标危害重要的器官,如肝、肾等。偶氮化合物在体内分解,可形成芳香胺化合物,芳香胺在体内经过代谢活动后与靶细胞作用而可能引起癌肿[3]。该染料如果在食品加工过程中使用就会污染食品成品,食用后可能会引起食物中毒,如果在长期食用的情况下,有可能对将来的生育造成影响,如不孕或者畸形儿,对自身健康也有影响,其为中等毒性致癌化合物,因此严格禁止在食品中使用[4-5]。夏立娅等[6]在国内某些品牌的辣椒面产品中检测出非法添加的非食用色素酸性橙Ⅱ。
碱性嫩黄O是一种芳香胺类碱性工业染料,一般用于腈纶、人造纤维、皮革、纸等的染色和棉织品、丝织品的印花[7]。在中性及偏碱性条件下,碱性嫩黄O与蛋白质吸附作用较强,不易褪色,一些不法商人将其加入食品及饲料中以改变产品的色泽来增加经济收入:渔民以及部分个体商户将黄鱼和腐竹侵染在含有碱性嫩黄O的液体中,使黄鱼和腐竹外表美观,达到以次充鲜、抬高价格等作用[8-9]。部分糖果和饮料中添加碱性嫩黄O用于维护和改善食品的外观颜色,以迎合消费者达到盈利目的[10]。毒理学资料表明碱性嫩黄O对人体皮肤黏膜有轻度刺激的危害,人接触或者吸入碱性嫩黄O可引起结膜炎、皮炎和上呼吸道刺激等症状,严重则引起中毒,长期接触甚至会致畸致癌[11],因此被严禁作为食品添加剂使用[12]。
目前,国内外检测食品中酸性橙Ⅱ和碱性嫩黄O的方法主要有薄层色谱扫描法、示差分析法、超高效液相色谱-质谱法(ultra-performance liquid chromatography-mass spectrometry,UPLC-MS)、气相色谱-质谱-离子扫描联用(gaschromatography-massspectrometry/selectedionscan,GCMS/SIM)、液相色谱串联质谱(LC-MS/MS)等。薄层分离-紫外吸收分光光度法设备简单,可操作性强,但是灵敏度不高[13];UPLC-MS、GC-MS/SIM、LC-MS/MS这类方法虽然分析时间短,而且灵敏度高,检出限低,但是需要昂贵的液相色谱及高分辨率的质谱仪,而且这类方法对实验操作人员要求较高,并不适合普及性的大面积使用[14-15];电化学方法通常设备简单,环境污染小,但是此类方法不易区分结构类似物[16]。近年来随着高效液相色谱仪的普及,并且它本身具有灵敏度高,分辨率高,检测结果精确的优点,而被广泛的应用于分析检测实验中。现有的高效液相色谱法检测食品中酸性橙Ⅱ和碱性嫩黄O的方法多采用梯度洗脱,分段波长的检测方法[17-18],这类方法操作复杂,仪器设备要求高,实验花费大[19],不适应方法的普及和日常监管。
本研究的目的是建立在“等梯度,恒定波长”的条件下同时快速检测食品中酸性橙Ⅱ、碱性嫩黄O这2类非食用色素的高效液相色谱检测方法,相较于现阶段通用的梯度洗脱,分段波长的检测方法分析更快速准确,实际操作更简单,对实验人员要求和仪器条件要求更低,同时本研究对建立的液相检测条件进行优化,主要包括波长、柱温、流动相和流速等,以探讨出结果可靠、灵敏度高、精密度高的最佳液相检测条件。
1 材料与方法
1.1料与试剂
酸性橙Ⅱ标准品、碱性嫩黄O标准品:上海金穗生物科技有限公司;甲醇:上海科顿企业技术咨询有限公司;乙腈(色谱纯)、甲酸(分析纯)、无水乙醇(分析纯)、乙酸铵(优级纯):成都市科龙化工试剂厂。本实验所测定食品样品均购于四川省雅安市苍抨路菜市场。
1.2器与设备
Shimadzu10A高效液相色谱仪:日本岛津公司;UV-3000紫外可见分光光度计:上海美谱达仪器有限公司;M illipore M illi-Q型纯水仪:美国M illipore公司;Sartorius CP225D型电子天平:德国Sartorius公司。
1.3验方法
1.3.1佳吸收波长
用紫外可见分光光度计分别对0.1 g/L碱性嫩黄O标准溶液、0.1 g/L酸性橙Ⅱ标准溶液、20mg/L酸性橙Ⅱ和碱性嫩黄O的混合标准溶液进行波段扫描(400~700 nm),可知碱性嫩黄O和酸性橙Ⅱ的各自最佳吸收波段,酸性橙Ⅱ和碱性嫩黄O的混合标准溶液的最大吸收波长,根据两种非食用色素的紫外吸收曲线,及混合物的最大吸收波长初步确定色谱检测的最佳吸收波长X0nm。
1.3.2动相的选择
参考林赛君等[20]的研究结果,实验初步选择的流动相为以下3种:甲醇-20mmol/L乙酸铵水溶液=75∶25、70∶30、65∶35、60∶40、50∶50(V/V),甲醇-50 mmol/L乙酸铵水溶液=75∶25、70∶30、65∶35、60∶40、55∶45、50∶50(V/V),乙腈-10 mmol/L乙酸铵水溶液=75∶25、70∶30、65∶35、60∶40、55∶45(V/V)。
在流速1.0m L/m in、检测波长X0nm、柱温35℃的条件下,准确注入10μL的混合标准品溶液,分别采用上述3种流动相的不同比例对标准溶液作等度洗脱,分析混合标准品的色谱图,确定流动相的最佳检测体积比例。
1.3.3温的选择
在流速1.0m L/min,进样量10μL,检测波长X0nm的条件下,按照确定的流动相试验,准确注入10μL的混合标准品溶液,分别在25℃、30℃、35℃、40℃、45℃柱温条件下,分析混合标准品的色谱图,确定检测此两种物质的最佳柱温。
1.3.4速的选择
在进样量10μL,检测波长X0nm,检测温度T的条件下,按照确定的流动相试验,准确注入10μL的混合标准品溶液,分别在1.0m L/min、0.9m L/min、0.8m L/min、0.7m L/min的流速条件下,分析混合标准品的色谱图,确定检测此两种物质的最佳流速。
1.3.5测固定波长的确定
以上述实验的结果来确定柱温、流动相等条件,流动相流速为1.0m L/min,在此系统下准确注入10μL的混合标准品溶液,实验检测波长分别为X0-15 nm、X0-10 nm、X0-5 nm、X0nm、X0+5 nm、X0+10 nm、X0+15 nm的条件下,分析混合标准品的色谱图,以两种非食用色素总的峰面积大小作为评价指标,并综合考虑单色素的蜂面积大小,最终确定检测此两种物质的最佳波长。
2 结果与分析
2.1佳吸收波长的初选
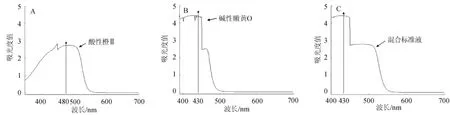
图1 碱性嫩黄O标准溶液(A),酸性橙Ⅱ标准溶液(B)及混标(C)的紫外吸收扫描结果Fig.1 UV absorption scanning results of auramine O(A),acid orange II(B)standard so lution and m ixed standard solution(C)
用紫外可见分光光度计分别对0.1 g/L碱性嫩黄O标准溶液、0.1 g/L酸性橙Ⅱ标准溶液、20mg/L酸性橙Ⅱ和碱性嫩黄O的混合标准溶液进行波段扫描,结果如图1所示。由图1可知,碱性嫩黄O在波长430 nm处吸光度值最高,酸性橙Ⅱ在波长480 nm处吸光度值最高,混合标准溶液在波长430 nm处达到最高,但是碱性嫩黄O以及混合标准溶液在波长450 nm之后吸光度值落差较大,综合考虑,初选450 nm为检测波长。
2.2动相的优化
选用Kromasil C18(4.6mm×150mm,5μm)色谱柱,在流速为1.0m L/min,检测波长为450 nm,柱温为30℃的条件下对混合标准溶液在不同流动相及比例下进行高效液相色谱检测。本实验主要选择了以下流动相及比例进行试验∶甲醇-20mmol/L乙酸铵水溶液=75∶25、70∶30、65∶35、60∶40、50∶50(V/V),甲醇-50mmol/L乙酸铵水溶液=75∶25、70∶30、65∶35、60∶40、55∶45、50∶50(V/V),乙腈-10mmol/L乙酸铵水溶液=75∶25、70∶30、65∶35、60∶40、55∶45(V/V),结果如图2所示。
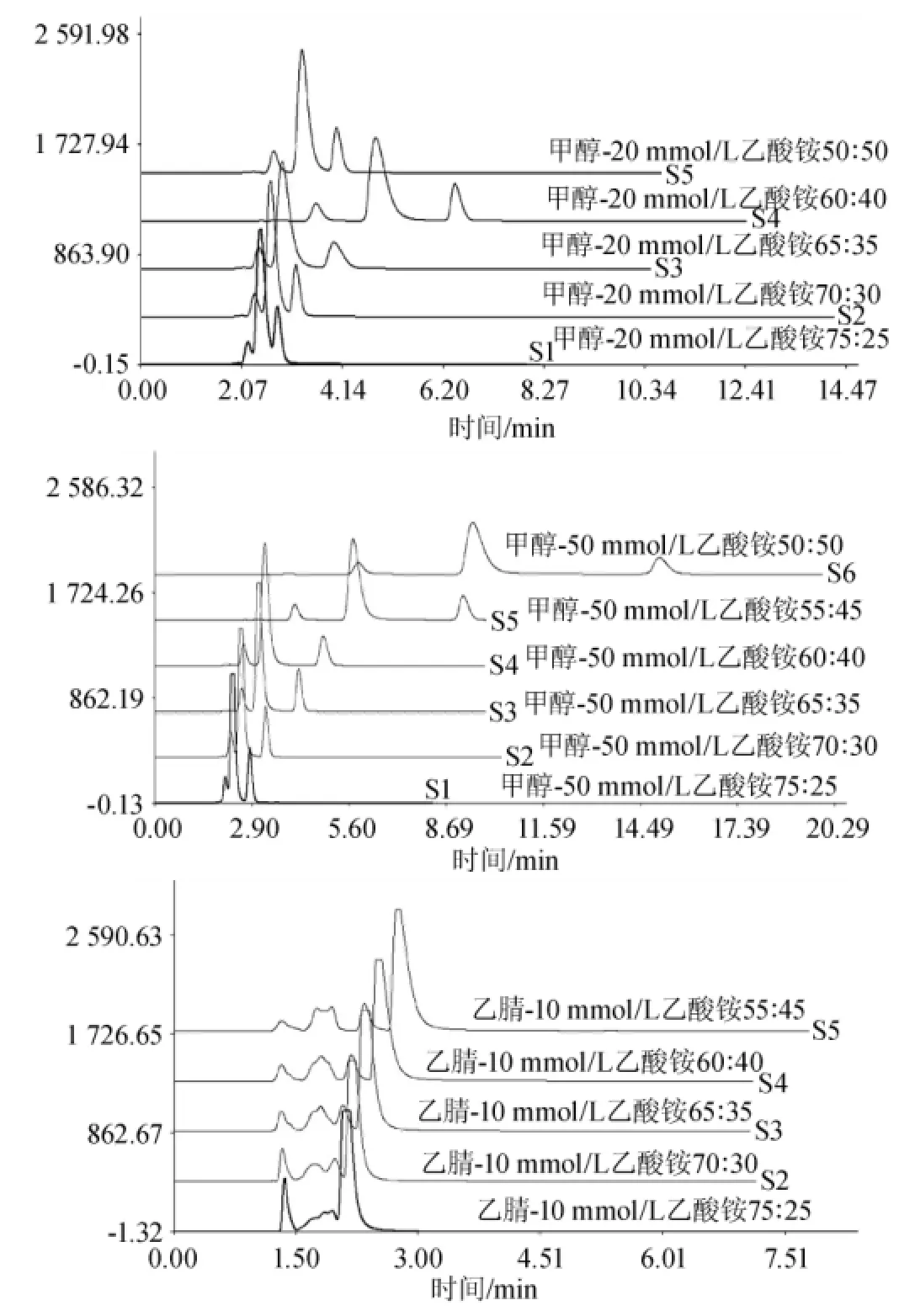
图2 混合标准溶液在不同流动相及比例条件下的高效液相色谱图Fig.2 HPLC chromatogram ofm ixed standard so lution under the conditions of differentmobile phase and proportion
由图2可知,当选用甲醇-20mmol/L乙酸铵水溶液为流动相时,调节流动相比例,酸性橙Ⅱ和碱性嫩黄O无法有效分离;当选用乙腈-10mmol/L乙酸铵水溶液为流动相时,碱性嫩黄O标准品溶液中的杂质会形成多个有效峰,杂质峰与碱性嫩黄O的峰分离难度增加,而且在此流动相条件下,酸性橙Ⅱ的峰提前,与杂质峰连接在一起,导致酸性橙Ⅱ与杂质峰也难以分离,且酸性橙Ⅱ出峰时间太早,易导致其与样品的杂质峰混合,因此该流动相不考虑;当选用甲醇-50mmol/L乙酸铵水溶液为流动相时,通过调节流动相比例,发现当甲醇-50 mmol/L乙酸铵水溶液= 55∶45(V/V)和甲醇-50mmol/L乙酸铵水溶液=50∶50(V/V)时,标准品杂质峰与酸性橙Ⅱ和碱性嫩黄O可以有效分离,但是因为在流动相为甲醇-50 mmol/L乙酸铵水溶液= 55∶45(V/V)时,酸性橙Ⅱ和碱性嫩黄O出峰时间相对较早,易导致样品杂质峰与目标峰的重叠,因此选用流动相为甲醇-50mmol/L乙酸铵水溶液=50∶50(V/V),在此条件下,混合标准溶液的高效液相色谱检测结果见图3。由图3可知,碱性嫩黄O和酸性橙Ⅱ的理论塔板数分别为3021和9084,峰底宽分别为0.477和0.435,分离度分别为5.62和7.11,且峰型正常,无拖尾现象。
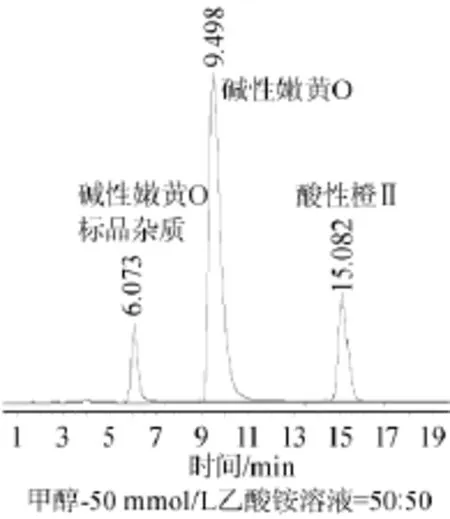
图3 最佳流动相条件下混合标准品溶液高效液相色谱图Fig.3 HPLC ch rom atogram ofm ixed standard solution under the optimalmobile phase conditions
2.3温的优化
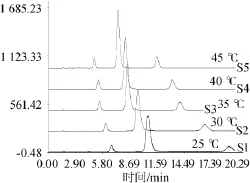
图4 不同柱温条件下混合标准品溶液高效液相色谱图Fig.4 HPLC chromatogram ofm ixed standard solution under different column tem perature conditions
在KromasilC18(4.6mm×150mm,5μm)色谱柱,流速1.0m L/m in,检测波长450 nm,流动相甲醇-50mmol/L乙酸铵水溶液的固定条件下,调节柱温(25℃、30℃、35℃、40℃、45℃)分别进行实验,结果如图4所示。由图4可知,随着柱温的增加,碱性嫩黄O和酸性橙Ⅱ的出峰时间明显提前,通过计算数据可知,在35℃时两种非食用色素总峰面积最大,且此时各峰保留时间合适。综合考虑对仪器的保护作用等因素,确定后续实验柱温为35℃。
2.4速的优化
在KromasilC18(4.6mm×150mm,5μm)色谱柱,柱温35℃,检测波长450 nm,流动相甲醇-50 mmol/L乙酸铵水溶液的固定条件下,调节流速(1.0m L/min、0.9m L/min、0.8m L/m in、0.7m L/min)分别进行实验,结果如图5所示。由图5可知,流速越小,相同物质出峰时间越长,流速越大,相同物质出峰时间越短;当流速为1.0m L/m in时,目标峰与杂质峰已能完全分离,且不易与样品中杂质峰叠加,因此选择流速为1.0m L/min。
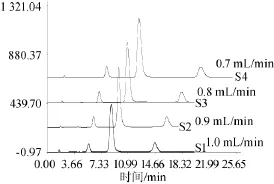
图5 不同流速条件下混合标准品溶液高效液相色谱图Fig.5 HPLC chrom atogram ofm ixed standard solution under different flow rates conditions
2.5测固定波长的确定
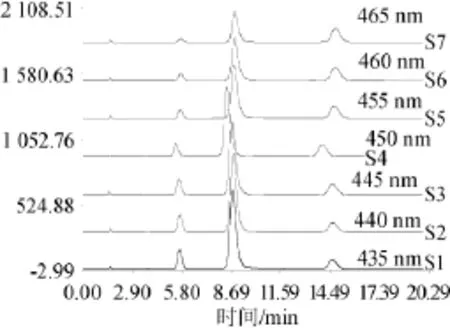
图6 不同波长条件下混合标准品溶液高效液相色谱图Fig.6 HPLC chromatogram ofm ixed standard solution under differentwave length conditions
在KromasilC18(4.6mm×150mm,5μm)色谱柱,柱温35℃,流速1.0m L/m in流动相甲醇-50mmol/L乙酸铵水溶液的条件下调节检测波长(435 nm、440 nm、445 nm、450 nm、455 nm、460 nm、465 nm)分别进行实验,结果如图6所示。由图6可知,随着波长的增加,碱性嫩黄O和酸性橙Ⅱ的总峰面积不断减少,但是酸性橙Ⅱ的峰面积不断增加,考虑到酸性橙Ⅱ的响应值较低,因此在满足两种非食用色素总峰面积最大的同时,也应尽量满足酸性橙Ⅱ的峰面积较大。由数据可观察出,碱性嫩黄O在450 nm之后,峰面积减少幅度较大,总峰面积减少幅度也较大,综合考虑两种非食用色素的总峰面积以及单色素的峰面积,选用检测波长为450 nm,在此条件下,两色素总峰面积较大,单色素的峰面积也较大。
2.6佳色谱条件的确定
综合上述试验结果,得到最佳的液相色谱条件为Kromasil C18(4.6mm×150mm,5μm)色谱柱、35℃柱温、1.0m L/min流速、450 nm波长、甲醇:50 mmol/L乙酸铵= 50∶50(V/V)为流动相。最佳条件下混合标准品高效液相色谱图见图7。由图7可知,两种非食用色素的保留时间、半峰宽、理论塔板数、分离度、拖尾因子、不对称度等都达到最佳,且此时仪器对两种非食用色素的相对吸收峰高都较好。
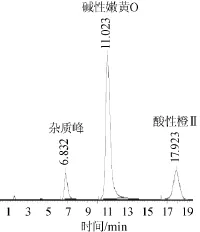
图7 最佳条件下混合标准品高效液相色谱图Fig.7 HPLC chromatogram ofm ixed standard solution under the optimum conditions
2.7品验证试验
采用本方法对市售以及超市的豆制品(包括腐竹、豆干等)、肉制品(鸡爪、真空包装鸡肉等)、鲜活类制品(黄鱼、真空包装黄鱼等)、调味品(包括辣椒粉、豆瓣酱等)共40份样品(如图8)中的非食用色素进行了检测,结果见图8。由图8可知,辣椒粉样品中检测到酸性橙Ⅱ含量为5.34mg/kg,小黄鱼样品中检测到碱性嫩黄O含量分别为1.92mg/kg,其余样品中未检测出非食用色素。
分析结果表明,建立的HPLC同时测定食品中酸性橙Ⅱ和碱性嫩黄O方法在12min内即可完成2种非食用色素的同时检测。本方法具有快速、简便、实用性强等特点。
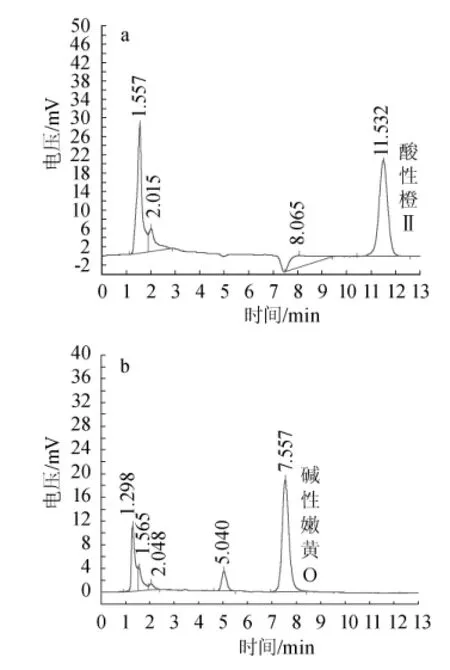
图8 辣椒粉(a)及黄鱼(b)的HPLC色谱图Fig.8 HPLC Chrom atog ram of chilli(a)and yellow c roaker(b)
3 结论
本试验采用等梯度、定波长的方法来分离检测食品中酸性橙Ⅱ和碱性嫩黄O,建立方法过程中由于仪器与实验方法改变的原因,初始阶段尝试了多种流动相的选择,建立了同时检测酸性橙Ⅱ和碱性嫩黄O这2种非食用色素的方法后并对该方法进行了优化,优化后的检测条件为:色谱柱为Kromasil C18(4.6mm×150mm,5μm);流动相为甲醇-50mmol/L乙酸铵溶液=50∶50(V/V);流速:1.0m L/m in;检测波长450 nm;柱温35℃。在上述条件下,碱性嫩黄O的保留时间为7.632m in,酸性橙Ⅱ的保留时间为11.548m in,碱性嫩黄O和酸性橙Ⅱ的理论塔板数分别为3021和9084,峰底宽分别为0.477和0.435,分离度分别为5.62和7.11,且峰型正常,无拖尾现象。该方法下样品重现性好,能在较短时间内完成2种物质的分离检测,方法简便易行。本方法比现有的研究方法操作更为简便,对仪器和操作人员的要求低,精密度高,具有良好的分离度,测定结果可靠,灵敏度高,能满足食品中酸性橙Ⅱ和碱性嫩黄O的含量测定。
随着对食品中非食用色素不断深入的研究,目前关于利用高效液相色谱检测食品中酸性橙Ⅱ和碱性嫩黄O的方法也是多种多样。本研究用高效液相色谱仪在等梯度、定波长的条件下分离并检测食品中的这2种非食用色素,可满足于市场上一般食品中酸性橙Ⅱ和碱性嫩黄O的检测。而且该方法更简单、快捷,可在12min内完成2种非食用色素的检测,且灵敏度高,适合推广使用。
[1]于立青,彭蜀晋.食品色素和人体健康[J].化学教育,2005,26(6):3-5.
[2]AM IN K A,HAMEID A,ABD ELSTTAR A H.Effectof food azo dyes tartrazine and carmoisine on biochemical parameters related to renal,hepatic function and oxidative stress biomarkers in youngmale rats[J].F Chem Toxic,2010,48(10):2994-2999.
[3]刘成伦,李小庆,王晶,等.偶氮类非食用色素的快速测定方法研究进展[J].食品科学,2009,30(5):273-276.
[4]严昌武,段晋超.1999~2003年绵阳市市售食品中非法使用酸性橙Ⅱ的调查[J].中国食品卫生杂志,2004,16(6):538-539.
[5]卢士英,邹明强.食品中常见的非食用色素的危害与检测[J].中国仪器仪表,2009(8):45-50.
[6]夏立娅,吴广臣,匡林鹤,等.辣椒面中非食用着色剂酸性Ⅱ的定量和定性检测的研究[J].分析检测,2007(12):178-180.
[7]郑小严.超高效液相色谱-串联质谱法同时测定食品中碱性橙、碱性嫩黄O和碱性桃红T[J].分析科学学报,2009,25(4):409-413.
[8]王建伟,钟海娟,梁炽琼.固相萃取-超高效液相色谱串联质谱法测定食品中碱性橙、碱性嫩黄O[J].分析测试技术与仪器,2010,16(2):108-112.
[9]冯济富,黄明.一起使用碱性橙浸染小黄鱼案的评析[J].中国卫生监督杂志,2003,10(6):351-352.
[10]YOSHIOKA,ICHIHASHI.Determ ination of 40 synthetic food colors in drinks and candies by high-performance liquid chromatography using a short column w ith photodiodearray detection[J].Talanta,2008,74(5): 1408-1413.
[11]高洁,尹峰,何国亮,等.高效液相色谱法测定豆制品中的碱性嫩黄O[J].分析试验室,2008(27):230-232.
[12]中华人民共和国卫生部.GB 2760—2011食品添加剂使用标准[S].北京:中国标准出版社,2011.
[13]刘慧,胡仰栋,孙小云.连续波长紫外分光光度法对合成食用色素混合体系的同时定量测定[J].食品工业科技,2006,27(9):164-166.
[14]林黛琴,万承波,邱萍,等.液相色谱-串联质谱法快速测定食品中4种黄色工业染料[J].质谱学报,2013,34(3):170-178.
[15]王翔.N-亚硝胺化合物和非法食品添加剂碱性橙Ⅱ的GC/MS检测方法研究[D].上海:复旦大学硕士论文,2009.
[16]GRYGART,KUCKOVA S,HRADILD,etal.Electrochemicalanalysis of natural solid organic dyes and pigments[J].J Solid State Electr,2003,7(10):706-713.
[17]李晶,李雪梅,徐济仓,等.超高效液相色谱法快速分离测定糖精钠、芝麻酚、酸性橙Ⅱ和碱性嫩黄O[J].光谱实验室,2013,30(3):63-69.
[18]谭建林,苏钟璧,王璐,等.高效液相色谱法同时测定食品包装材料中9种禁用染料的含量[J].安徽农业科学,2012,40(16):8954-8966.
[19]赵榕,赵海燕,李兵,等.建立同时测定调味品中非法添加的4种工业染料的SPE-UPLC-MS/MS法研究[J].中国食品卫生杂志,2009,21(5):410-414.
[20]林赛君,屠海云,肖海龙,等.高效液相色谱-串联质谱法同时测定食品中五种黄色化工染料[J].色谱,2011,29(1):79-82.
Optim ization of simultaneousdetection ofacid orange IIand auramineO in food by HPLC
A method of simultaneous detection of acid orange IIand auram ine O in food by HPLC wasestablished.The influence factors including chromatographic column,column tem perature,the type and proportion ofmobile phase,flow rate and wavelength were researched and optimized. The chromatographic detection condition was obtained as follow s:chromatographic column K romasil C18(4.6 mm×150 mm,5μm),methanol -50mmol/L ammonium acetate(50∶50,V/V)asmobile phase,flow rate 1.0m l/min,determ inewavelength 450 nm,and column temperature 35℃. Under the optimum conditions,auram ine O and acid orange IIhad good separation degree and normal peak type.The resulted showed that the methodwassuitable fordetection ofauramineO and acid orange IIin food.
food;acid orange II;auramineO;HPLC
O657.7
0254-5071(2016)01-0165-05
10.11882/j.issn.0254-5071.2016.01.037
LISha1,2,LEIZhiqi2,ZHU Hong2,ZHANGZhiqing1*
(1.College ofFood Science,Sichuan AgriculturalUniversity,Ya’an 625014,China;2.Chongqing Fuling Institute for Food and Drug Control,Chongqing 408000,China)
2015-11-17
四川农业大学校县合作项目(0600H0300)
李莎(1990-),女,硕士研究生,研究方向为食品加工与安全。
张志清(1976-),男,博士,教授,研究方向为粮油副产物开发利用及食品检验。
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:28
食品安全导刊(2020年21期)2020-09-07 09:14:04
农家科技中旬版(2019年9期)2019-10-08 05:27:47
山西农业科学(2019年6期)2019-06-19 07:14:40
福建基础教育研究(2019年8期)2019-05-28 08:39:51
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:43
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:45
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
山东工业技术(2016年13期)2016-06-29 09:05:13
浙江农业科学(2016年11期)2016-05-04 04:16:45
