
仙茅配方颗粒质量标准研究
2016-09-16 07:17谢鸣坤郑智慧梁华伦徐万帮
中国中医药现代远程教育 2016年13期
谢鸣坤 郑智慧 梁华伦 徐万帮
(1 广东省中医院药剂科,广州 510120;2 广东省食品药品检验所,广州 510120)
仙茅配方颗粒质量标准研究
谢鸣坤1郑智慧1梁华伦1徐万帮2*
(1广东省中医院药剂科,广州510120;2 广东省食品药品检验所,广州510120)
目的 建立仙茅配方颗粒的质量标准。方法 运用薄层色谱法,以仙茅苷为对照,建立仙茅配方颗粒的薄层鉴别方法。采用高效液相色谱法建立仙茅配方颗粒仙茅苷的含量测定方法。结果 薄层鉴别斑点清晰,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。高效液相色谱法测定仙茅苷在0.1404~2.808 μg/ml的范围内呈良好线性关系(r=0.9998),加样回收率在97.3%~103.4%,RSD为1.9%(n=6)。结论 该方法操作简便,结果准确,重现性好,可用于仙茅配方颗粒的质量控制方法。
仙茅配方颗粒;质量标准;薄层色谱法;高效液相色谱法
中药配方颗粒是指运用现代药物加工工艺对传统中药材的关键成分进行提取、分离以及浓缩,干燥后将有效成分制成颗粒状,可供使用者直接冲服的颗粒[1]。中药配方颗粒在保持原有药物功效及作用的前提下,实现了药物便捷使用、顺利服用和高效作用,因此,临床应用日益广泛[2-4]。基于此,国家食品药品监督管理总局为了加强对中药配方颗粒的管理,引导该产业的健康发展,在2015年12月24日颁布了《中药配方颗粒管理办法》[5],在该办法实施后,相信中药配方颗粒将迎来跨越式的发展。
仙茅为石蒜科多年生草本植物仙茅的干燥根茎[6]。其味辛,性热、有毒。具有消散痈肿、补肾阳、强筋骨、祛寒湿、益精血等功效。常用于痈疽肿痛、肾阳不足、阳痿精冷、筋骨酸软、腰膝冷痹、阳虚冷泻等症[7]。仙茅的应用极为广泛,是常用中药材之一,本文参照《中国药典》仙茅项下的要求,对仙茅配方颗粒的质量标准进行研究,旨在为仙茅配方颗粒的质量控制提供技术支撑。
1 仪器和试剂
1.1仪器 仪器薄层色谱自动点样仪 (CAMAG AUTOMATIC TLC SAMPLER4)、薄层自动成像仪 (CAMAG REPROSTAR 3),预制硅胶G薄层板,购于Merck公司、青岛海洋化工厂分厂、烟台市化学工业研究所及国药集团化学试剂有限公司;其它试剂均为分析纯。高效液相色谱仪:Aglient 1260,SHIMADZU(LC-20AT);色谱柱:资生堂 MGⅡ(5 μm,150 mm×4.6 mm),phenomenex luna100A(5 μm,150 mm×4.6 mm)。电子天平:Sartorius-CP225D,超声波清洗仪: E-ELMA (TI-H15 MF3)。
1.2试剂 仙茅苷对照品,购于中国药品生物制品检定所 (供含量测定用,编号110771-201105,含量为89.2%)、仙茅配方颗粒 (自编1,2,3);仙茅配方颗粒阴性(仙茅配方颗粒辅料麦芽糊精组成);乙腈为色谱纯;水为实验室自制高纯水,其余试剂均为分析纯。
2 实验方法
2.1薄层鉴别 参照《中国药典》2010年版一部94页仙茅【鉴别】项下的方法,稍微修订,在此基础上制定了以仙茅苷对照品为对照的仙茅配方颗粒的薄层色谱鉴别方法,具体操作如下:取本品适量,研细,取0.5 g,加乙醇20 ml,超声处理20 min,滤过,滤液蒸干,残渣加乙酸乙酯1 ml使溶解,取上清液作为供试品溶液。另取仙茅苷对照品,加乙酸乙酯制成每1 ml含0.1 mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,吸取上述两种溶液各2 μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲醇-甲酸(10∶1∶0.1)为展开剂,展开,取出,晾干,喷以2%铁氰化钾溶液-2%三氯化铁溶液(1∶1)的混合溶液。
2.2检查项 水分、溶化性、粒度、浸出物(按热浸法,以乙醇为溶剂,进行提取)等参照中国药典2010年版一部附录有关颗粒剂项下执行。
2.3含量测定 参照《中国药典》2010年版一部94页仙茅含量测定方法,制定仙茅配方颗粒中仙茅苷的含量测定方法,经方法学验证,认为该方法基本可行。
2.3.1色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%磷酸溶液(21∶79)流动相;检测波长为285 nm。理论板数按仙茅苷峰计算应不低于3000。
2.3.2对照品溶液的制备 将仙茅苷置五氧化二磷真空干燥箱中,室温干燥过夜。取仙茅苷对照品适量,精密称定,加甲醇制成每1 ml含0.1 mg的溶液,摇匀,备用。
2.3.3供试品溶液的制备 取本品适量,研细,取约1 g,精密称定,置具塞锥形瓶中,精密加入甲醇20 ml,密塞,称定重量,超声处理(功率200 W,频率50 kHz)30分钟,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液、摇匀备用。
2.3.4阴性溶液的制备 取阴性样品适量,研细,取约1 g,同2.2.3,制备阴性溶液
3 结果与讨论
3.1薄层鉴别 参照2.1的实验步骤,在展开缸中饱和15 min后,在室温条件下展开(T=25℃,RH=68%),当展开至约9 cm时取出,晾干,喷以2%铁氰化钾溶液-2%三氯化铁溶液(1∶1)的混合溶液。此时,在与对照品色谱相应的位置上,显相同的蓝色斑点,色谱图如下,见图1。

图1 仙茅配方颗粒薄层鉴别色谱图
由图1可知,薄层色谱展开斑点分离度较好,Rf在0.3~0.8之间,阴性对照无干扰,因此,方法可行。
同时,由于该方法直接沿用《中国药典》 (一部)2010年版94页所记载方法,因此只对其适应性做考察,不再做方法学验证。
3.2仙茅配方颗粒检查
3.2.1水分 按照《中国药典》2010年版附录IXH水分测定项中烘干法测定。取仙茅配方颗粒(自编1),平行2份,测得平均含水量为3.6%,符合2010版药典一部中关于颗粒剂的要求。
3.2.2溶化性 取仙茅配方颗粒一包,加热水200 mL搅拌5 min,观察到颗粒全部溶化,无焦屑等异物,说明该颗粒的溶化性符合要求。
3.2.3粒度 按照《中国药典》2010年版一部附录XI B粒度测定第二法,取仙茅配方颗粒30 g,称定质量,置规定的药筛中,保持水平状态过筛,左右往返,筛动3 min,不能通过一号筛和能通过五号筛的颗粒为1.93 g(不超过巧%)。
3.2.4浸出物 参照《中国药典》附录要求,采用乙醇为溶剂,用热浸法,测得三份浸出物的数据如下

表1 仙茅配方颗粒浸出物
从表1可以看出,所得三批仙茅苷的浸出物的平均值均接近30%,比仙茅药材的浸出物高。
3.3仙茅苷含量测定
3.3.1系统适用性试验(专属性试验)参照《中国药典》2010年版一部94页仙茅含量测定方法和2.3的实验步骤,取供试品,对照品和阴性溶液各10滋l注入液相色谱仪,记录色谱图。如图2、3、4。

图2 仙茅苷对照品图谱
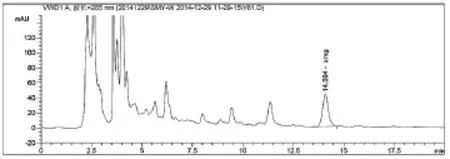
图3 仙茅配方颗粒(自编3)图谱
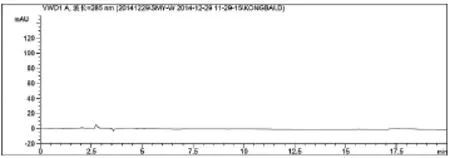
图4 仙茅阴性对照图谱
从图2~3可以看出,仙茅苷和样品 (自编3)中其它相邻组分色谱峰可达到基线分离,仙茅苷与相邻色谱峰的分离度大于1.5,按仙茅苷峰计算,此时理论板数为11771,大于3000,符合《中国药典》所规定的要求。
3.2.2提取方法的考察 取经粉碎后的本品约1.0 g(自编1),采取超声、冷浸、回流不同提取方式,按2.3.3的处理方式,分别提取60 min。精密吸取各供试液10滋l,注入液相色谱仪,按上述色谱条件测定,得峰面积,计算含量,结果见表2。

表2 不同提取方法的比较结果表
从表2可以看出,三种提取方式结果相差不大,但冷浸方式相对含量偏低,可能与冷浸过程中提取不完全有关,回流提取操作相对繁琐且能耗较大,而超声简便、迅速且无污染损失小,因此采用超声提取方法。
3.2.3提取时间的考察 取经粉碎后本品1.0 g(自编1),精密称定,照2.3.3的处理方式,超声不同时间。精密吸取各供试液10滋l,注入液相色谱仪,按上述色谱条件测定,得峰面积,计算含量,结果见表3。

表3 不同提取时间的比较结果表
从表3可以看出,超声30 min后,仙茅苷的有效成分基本提取完毕,因此,本质量标准选择超声30 min为提取时间。
3.2.4提取溶剂的考察 取经粉碎后本品1.0 g(自编1),精密称定,照2.3.3的处理方式,分别精密加入70%的甲醇、乙醇和甲醇20 ml,并分别用相应的溶液补足减失重量。精密吸取各供试液10滋l,注入液相色谱仪,按上述色谱条件测定,得峰面积,计算含量,结果见表4。

表4 不同提取溶剂的比较结果表
从表4可以看出不同溶剂提取结果基本一致,但在实验中发现乙醇提取的样品杂质峰相对较多,可能与仙茅中其他成分易溶于乙醇的物理性质有关,故本质量标准采用甲醇作为仙茅配方颗粒的提取溶剂。
3.2.5线性关系考察 分别精密吸取仙茅苷系列对照品溶液,注入液相色谱仪,测定峰面积,以仙茅苷的峰面积值(y)为纵坐标,以仙茅苷的进样量(x)为横坐标,计算得回归方程为y=556.8x+7.0876,r2= 0.9998,结果见表5。

表5 峰面积积分值与进样浓度的关系
从表5可知,仙茅苷进样量在0.10029~2.00571 mg/ ml范围内,进样量与峰面积值呈良好的线性关系。
3.2.6精密度 精密吸取仙茅苷对照品溶液((1.00286 mg/m)l10 μl,重复进样6次,测定仙茅苷峰面积积分值,计算RSD,结果见表6。
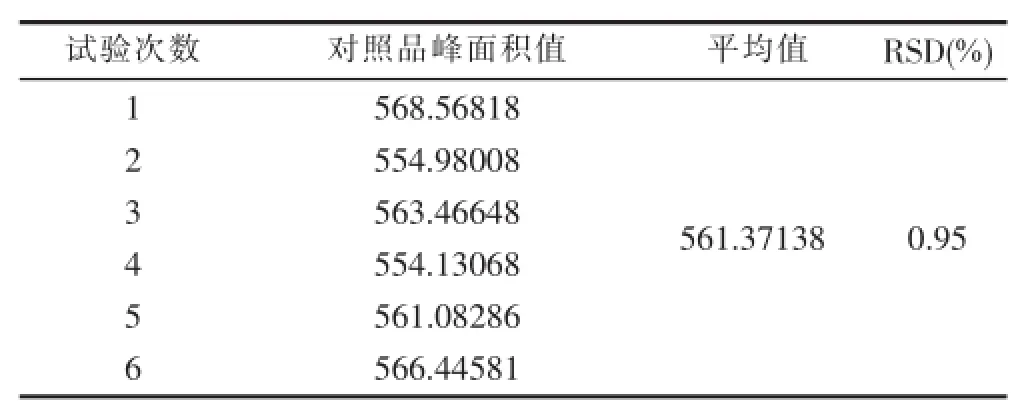
表6 精密度试验结果
从表6可以看出,采用上述方法,测定仙茅苷的峰面积,精密度良好,RSD为0.95%,小于2.0%,符合《中国药典》的要求。
3.2.7中间精密度的考察 为了考察不同操作人员在上述实验条件下对实验的影响,本实验分别由本实验室2名实验人员分别取样(自编1)1份,按2.3的操作。分别在安捷伦1260高效液相色谱仪和岛津LC-20AT上测定仙茅苷峰面积,计算含量,结果见下表7。
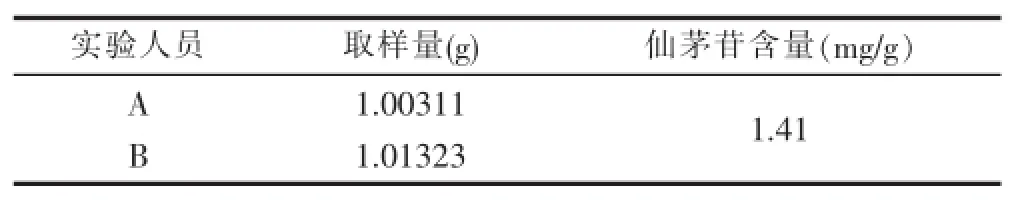
表7 中间精密度试验结果
从表7可以看出,该方法在不同实验者和不同仪器上测试结果稳定,可靠,说明该方法的中间精密度良好。
3.2.8日间精密度(稳定性试验)为了考察取同一批号样品(自编1),将供试品溶液放冰箱保存,每隔一段时间进样一次,共测定48 h,分别进样10μl,测定样品中仙茅苷峰面积积分值,计算RSD,结果见表8。

表8 稳定性试验结果表
从表8可以看出,仙茅苷在冰箱中保存,48 h内基本稳定,所测得峰面积大小值的RSD为0.97%,能满足质量控制的基本要求,不过,尽管仙茅苷在冰箱中保存比较稳定,仍建议大家在较短时间内尽快完成测试,以防其他因素引起仙茅苷的降解而导致不必要的误差。
3.2.9重复性试验 精密称取同一批号(自编1)仙茅配方颗粒,平行进样6次,记录仙茅苷峰面积积分值,计算含量,计算RSD,结果见表9。

表9 重现性试验结果表
从表9可以看出,仪器精密度良好,所测得的重复性良好,该批号仙茅配方颗粒中仙茅苷的平均含量为1.37 mg/g,其中RSD为0.5%。
3.2.10回收率试验 取已知含量的样品(自编1,含量为1.37 mg/g)0.5 g,精密称定,共6份,精密称定,分别精密加入仙茅苷对照品溶液(0.10029 mg/m)l7 ml,按正文供试品溶液制备方法和色谱条件,制备加样回收供试品溶液,注入液相色谱仪,测定,计算回收率,平均回收率,RSD,结果见表10。

表10 仙茅苷配方颗粒回收率试验
从表10可以看出,采用该方法所得平均回收率在97.3%~103.4%之间,RSD为1.9%,样品回收率良好。
3.2.11样品测定 取样品3批,每批约1.0 g,精密称定,按供试品溶液制备方法制成供试液,分别进样10 μl,测定峰面积,计算仙茅苷含量,结果见表11。
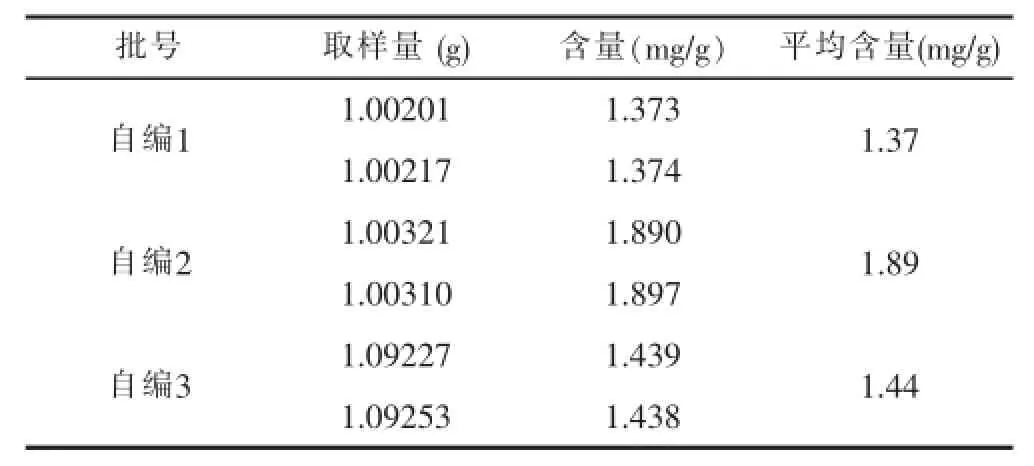
表11 样品测定结果
从表11可以看出,3批仙茅配方颗粒中仙茅苷的含量基本相近,所测试同一批号的2份样品结果接近。
4 结论
通过对上述实验可知,仙茅配方颗粒是通过对仙茅药材进行提取后,经多方面处理,加入麦芽糊精所制得的新剂型,具有携带方便,使用便利等诸多优点,其主要成分上和原材料基本一致。
在本质量标准的研究中,考察了薄层色谱,各检查项,同时建立了高效液相色谱法测定仙茅配方颗粒中仙茅苷含量的测定方法,通过方法学考察,方法可行,结果准确,可以为仙茅配方颗粒质量标准的建立提供技术支撑,但是,在本实验中所涉及的样品批次偏少,无法制定合理的限度标准。因此,在实际大规模生产中,有必要多积累数据,建立合理的限度标准,以期到达合理的质量控制。
[1]陈昭,毕晓黎,邱宏聪,等.中药配方颗粒剂质量控制研究现状及展望[J].中国民族民间医药,2013,22(20):11-13.
[2]王永和.中药配方颗粒的现状及最新研究进展[J]中国卫生产业,2014,(8):188-189.
[3]王金凤,张钦德.中药配方颗粒药效学研究进展[J]中国中医药信息杂志,2013,20(8):110-112.
[4]王乙同.中药配方颗粒剂的发展现状与未来趋势[J].大家健康,2014,8(13):325-327.
[5]国家食品药品监督管理总局关于征求《中药配方颗粒管理办法(征求意见稿)》意见的公告(2015年第283号)http://www.sda.gov.cn/WS01/CL1037/ 140101.html
[6]杨婉梅,杨永红.中国药用植物仙茅开发利用综述[J].现代农业科技,2015(8):110.
[7]杨光义,叶方.仙茅药理作用和临床应用研究概述[J].中国药师,2011,14(7):1039-1041.
Study on the Quality Standard of Rhizoma Curculigin is Formula Granule
XIE Mingkun1,ZHENG Zhihui1,LIANG Hualun1,XU Wanbang2
(1.Department of Pharmacy,Guangdong Provincial Hospital of TCM,Guangzhou 510120,China;2.Guangdong Institute of Drug Control,Guangzhou 510120,China)
Objective To establish a quality standard of Rhizoma curculiginis f ormula granule(RCFG).Methods A TLC identification method was established,and curculigoside by HPLC of RCFG was determined.Results TLC were clear,and the spots of the samples were in the same position as the control samples.High performance liquid chromatography was used to determine curculigoside.The linear relationship from 0.1404 to 2.808 μg/ml is good(r=0.9999).The average recovery rate was 100.40%,and RSD was 0.78% (n=6).Conclusion The method is simple,accurate and reproducible,and can be used for the quality control method of Rhizoma Curculigini.
Rhizoma Curculigini formula granule;quality standard;TLC;HPLC
10.3969/j.issn.1672-2779.2016.13.060
1672-2779(2016)-13-0130-05
wbxu@163.com
(本文编辑:李海燕 本文校对:黄桂红2016-04-15)
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
科学与财富(2021年35期)2021-05-10
食品安全导刊(2020年14期)2020-12-04
食品安全导刊·中旬刊(2020年5期)2020-06-04
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
中成药(2017年3期)2017-05-17
海峡科技与产业(2016年3期)2016-05-17
云南中医学院学报(2015年3期)2015-07-31
中国当代医药(2015年8期)2015-03-01
