
β位取代基对咔咯锰(V)氧配合物电子吸收光谱的影响
2016-09-09 09:35陈华彬章小慧龚丽珍徐志广刘海洋
物理化学学报 2016年8期
陈华彬 章小慧 龚丽珍 何 婧 许 旋 徐志广,* 刘海洋
(1华南师范大学化学与环境学院,教育部环境理论化学重点实验室,广州510006;2华南理工大学化学系,广州510641)
β位取代基对咔咯锰(V)氧配合物电子吸收光谱的影响
陈华彬1章小慧1龚丽珍1何婧1许旋1徐志广1,*刘海洋2,*
(1华南师范大学化学与环境学院,教育部环境理论化学重点实验室,广州510006;2华南理工大学化学系,广州510641)
采用含时密度泛函理论(TDDFT)和B3LYP方法对一系列β位咔咯锰(V)氧配合物的几何结构、前线轨道、自然键轨道和电子吸收光谱进行了计算。并探讨了β位取代基对咔咯锰(V)氧配合物电子吸收光谱的影响。结果显示β位取代基效应中的空间效应是能导致该体系电子吸收光谱Soret带和Q带红移。β位取代基的空间位阻大时,导致配合物的咔咯环产生严重扭曲,引起Soret带和Q带吸收峰的ΔE能隙减小,导致Soret带和Q带发生红移。
咔咯锰(V)氧配合物;电子吸收光谱;TDDFT;取代基
www.whxb.pku.edu.cn
1 引言
咔咯是具有18π电子结构的类卟啉大环化合物1,环中的三个氢原子容易失去形成三价阴离子配体,因其空腔小,能与金属离子形成稳定的高价咔咯金属配合物2。近几年来,由于咔咯合成方法的不断改进3-5,有关咔咯的研究越来越受到人们的关注,咔咯金属配合物在催化氧化、催化还原、光动力疗法、传感器和太阳能燃料电池等方面有着越来越大的应用6。咔咯环上中位和β位能被修饰,使其衍生物及金属配合物更加多样化,取代基的变化会使紫外-可光(UV-Vis)光谱发生显著变化。Ghosh等致力于合成和研究Cu7-13、Fe14、Ni11、Ga11、Ag10等咔咯金属配合物,并进一步从理论和实验上研究取代基效应对体系结构和性质的影响。研究发现,中位苯环连有不同的推、拉电子基团的咔咯铜配合物,它们的电子吸收光谱和电化学性质会随取代基的推拉电子效应而发生一定的改变11;尤其是当咔咯铜配合物的β位连接―CF3、―Br等拉电子能力和空间位阻都比较大的取代基时,其构型会产生一定程度的扭曲变成鞍型结构10,12,体系的电子吸收光谱发生明显红移,且取代基的拉电子性质越强,红移程度越大。电子吸收光谱红移的现象在金属卟啉配合物上已有报道15-17。咔咯锰(V)氧配合物是锰催化氧化的活性中间体18,对其电子结构、反应活性及光谱特性研究是重要的课题。文献报道咔咯锰(V)氧配合物在不同取代基修饰的情况下,Soret带和Q带特征吸收发生红移。为此,本文研究咔咯锰(V)氧配合物的电子吸收光谱性质,通过含时密度泛函理论(TDDFT)分别对发生在中位,尤其是β位连接不同推、拉电子取代基团的咔咯锰(V)氧配合物的电子光谱进行理论计算及讨论Soret带和Q带红移的原因。
2 计算方法
为考察取代基效应对咔咯锰(V)氧配合物电子光谱的影响,本文设计出在咔咯环上中位和β位引入吸电子基团(―C6F5、―C6H3F2、―Br、―F)和供电子基团(―C6H5、―C2H5)构成7个配合物,结构见图1。参考以往文献19,20发现B3LYP方法优化的结构接近实验值,因此采用密度泛函的B3LYP方法,运用混合基组进行计算,其中对C、N、O、F、Br、H采用6-31G基组,Mn采用LanL2DZ基组,优化计算连接不同取代基的咔咯锰(V)氧配合物的分子几何结构。咔咯锰(V)氧配合物的核磁共振实验显示呈低自旋,锰原子最外层两个d电子成对排布21-26,有关理论计算也显示咔咯锰(V)氧配合物单重态最稳定,因此所有分子是以单重态进行计算的。计算各化合物优化构型的振动频率,结果均没有虚频,表明分子构型稳定。在优化的结构基础上,以B3LYP方法运用含时密度泛函理论进行电子吸收光谱计算,计算中对C、N、O、F、Br、H采用6-31+G(d,p),Mn采用LanL2DZ基组,同时引入溶剂化计算。溶剂效应采用自洽场反应理论(SCRF)的极化连续模型(PCM),所选溶剂为二氯甲烷(ε=8.93)。计算配合物1-7的电子吸收光谱,考察它们的跃迁轨道、吸收波长和振子强度等性质。以上所有计算过程均使用Gaussian 0327程序完成。
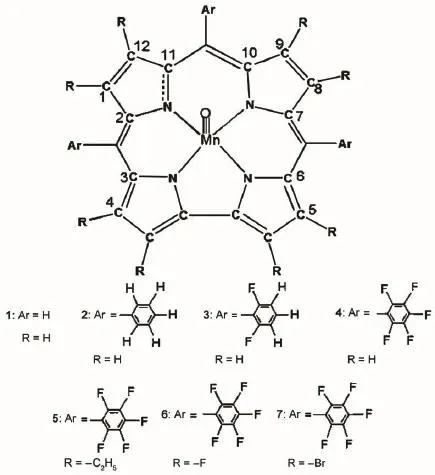
图1 咔咯锰(V)氧配合物的结构Fig.1 Structures of manganese(V)-oxo corrole complexes
3 结果与讨论
3.1配合物的几何结构
配合物1-7的几何优化结构见图2,结构数据列于表1。自由咔咯内腔的四个吡咯近似共面,由于锰原子与氧原子成键,锰原子偏离大环平面(见图2)。外围的取代基都为H的化合物1,其DN.N.N.N的角度接近零,四个吡咯的氮原子在一个平面上。中位上引入位阻更大的芳基后,DN.N.N.N没有多大的变化,二面角角度都小于1°;当β位连接位阻大的乙基、溴原子后,DN.N.N.N角度的数值增大到4°左右。观察上述分子的结构数据,咔咯环的结构变化不大,而dMn―O有稍微变化,变化主要集中在咔咯环的变形上。D1.2.3.4,D5.6.7.8,D9.10.11.12表示的是咔咯中两个吡咯间的二面角,1、3、4和6这4个化合物的3个二面角的绝对值几乎在10°以下,表明这几个化合物的咔咯骨架基本处于同一平面。而化合物2、5、7的二面角的绝对值比较大,其吡咯之间的二面角如,D1.2.3.4和D5.6.7.8的值在10°-25°左右,而结构变形更大的化合物5和7,其二面角D9.10.11.12增大至27°,变化尤为明显,这表明咔咯骨架存在严重的扭曲(见图2)。同时我们注意到,其中化合物5、7的DN.N.N.N的二面角在3.5°-5.0°之间(见表1),其角度略为增大。这主要因为化合物5、7的β位大位阻取代基团(―CH2CH3、―Br)与中位的芳基存在排斥作用,导致咔咯骨架发生扭曲。说明外围取代基的空间效应对咔咯环结构的影响明显,使咔咯环平面发生变形。
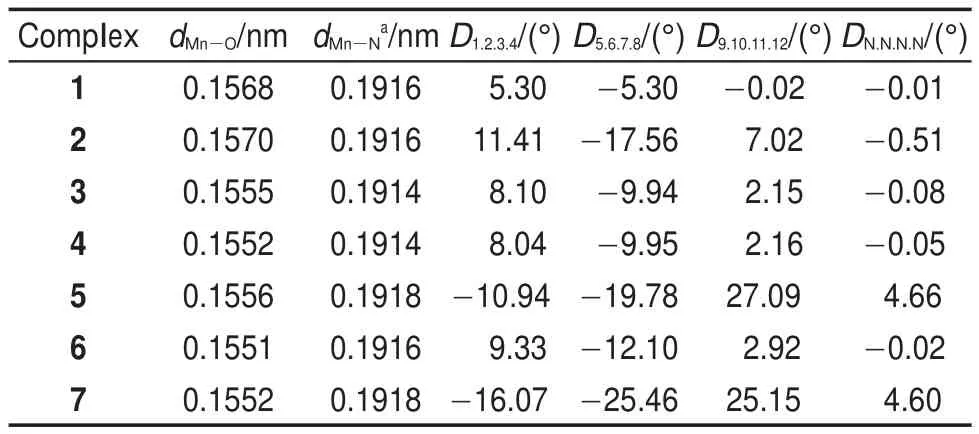
表1 配合物1-7的部分几何结构参数Table 1 Selected optimized geometrical structure parameters of 1-7 complexes
3.2自然布居分析(NPA)
表2给出配合物1-7的锰、氧原子和咔咯骨架的NPA电荷。数据显示,中位和β位引入不同取代基后(2-7和1比较),与取代基性质变化呈现规律性,中位取代基从吸电子性质从小到大排序为2、1、3、4,这与QCorrole带的负电大小顺序一致,分别为-0.5804、-0.5863、-0.6092、-0.6252e。另一方面,对于β位连接取代基的吸电子性质从小到大排序为5、4、7、6。该顺序也与QCorrole的负电荷大小完全一致。比较取代基电子效应与反映配合物结构变形程度的二面角数据,发现带供电子基团的配合物5和带吸电子基团的配合物7的咔咯环面均出现变形。这说明,取代基电子效应与配合物的咔咯环变形的关系不大。
3.3配合物的电子吸收光谱
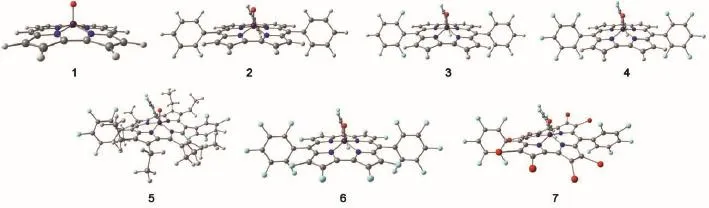
图2 配合物1-7的几何优化结构图Fig.2 Optimized geometrical structures of 1-7 complexes
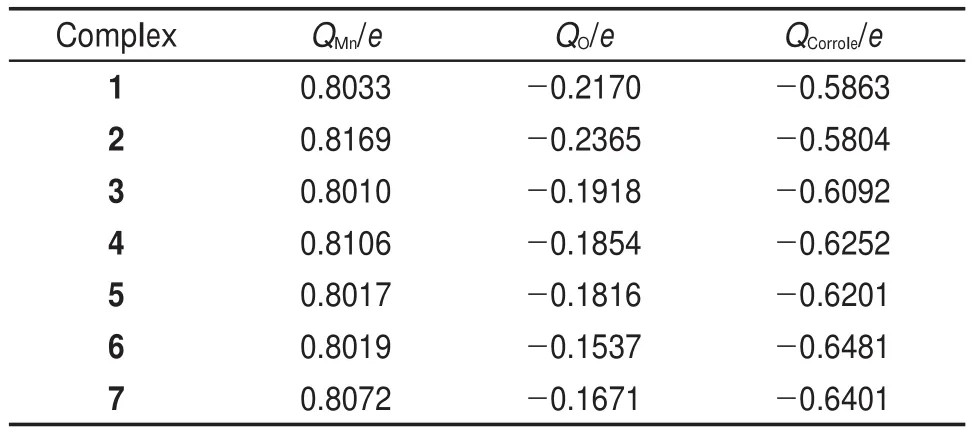
表2 配合物1-7的NPA电荷Table 2 Natural population analysis(NPA)charges of 1-7 complexes
Ghosh等13采用DFT方法对Cu、Ga、Ni等金属咔咯的电子结构进行研究,发现镓咔咯的电子吸收光谱与锌卟啉非常类似,可以采用“四轨模型”进行解释。“四轨模型”是由Gouterman等28,29在光谱测定的实验基础上运用分子轨道(MO)理论建立的,用于描述卟啉低激发光谱的Q带和B带。Q带和B带的跃迁主要涉及到a1u、a2u和eg轨道,a1u和a2u的能级高低由结构决定。B带也称为Soret带,是a2u-eg和a1u-eg通过组态相互作用产生。咔咯的紫外-可见光谱显示在350-450 nm附近有一个强烈的Soret吸收带,在480-650 nm区域则有较弱的Q吸收带。本文则采用含时密度泛函方法结合溶剂化模型进行电子吸收光谱计算,探讨咔咯锰(V)氧配合物在外围取代基的作用下,其Soret带和Q带发生的变化情况。
表3中列出在BP86、B3LYP和LSDA三种方法下通过计算得到的体系3、4、6和7的电子光谱数值。从该表中4个配合物的电子吸收光谱数据,我们发现在B3LYP方法下,计算光谱值与实验值最接近,因此我们采用B3LYP方法计算其余(1、2和5)的配合物(见表4)。
首先考察有相应实验值的3、4、6和7,具有非平面咔咯环的配合物7的3个吸收峰波长实验值明显大于另外三个咔咯环近似平面的3、4和6,表现出红移的现象。例如7中的Soret带波长为421 nm(计算值为428 nm),明显大于3的405 nm(395 nm)、4的404 nm(397 nm)和6的382 nm(393 nm)。另外,对于配合物3、4和6的实验值和计算值,其大小顺序出现的细微差异,可能是由于在不同溶剂测出的数据以及光谱本身灵敏度存在的偏差,但这偏差不影响β位取代基空间效应导致Soret带红移的讨论。结合以上数据说明扭曲的咔咯环结构可能是导致红移的原因。进一步考察其余3个化合物,发现存在咔咯环扭曲的配合物5的Soret带计算峰值为417 nm,明显大于配合物1和2 的380和399 nm,理论上预测应存在吸收峰红移现象。而且符合前述配合物7对比3、4、6的实验值呈现出红移的实验现象,说明咔咯环的扭曲构型应该是影响Soret带红移的主要因素。与此同时,表5提供7种配合物的Q带吸收光谱,各个配合物的Q带吸收峰在480-650 nm之间,符合咔咯配合物Q带吸收峰的归属。由于Q带受外围取代基的影响,从理论计算数据可以看出,配合物在外围取代基的作用情况下,Q带吸收峰值的变化情况大致与Soret带的红移变化情况类似。比如发生构型扭曲的配合物5和配合物7的Q带吸收峰分别为515 和535 nm(实验值为537 nm),也明显大于其他类平面结构的配合物,如配合物3的502 nm(518 nm)、4的505 nm(518 nm)、6的489 nm(503nm)。结合前述Soret带的红移特点,进一步表明本系列7个咔咯锰(V)氧配合物吸收峰发生红移与其结构发生扭曲有重要关系。
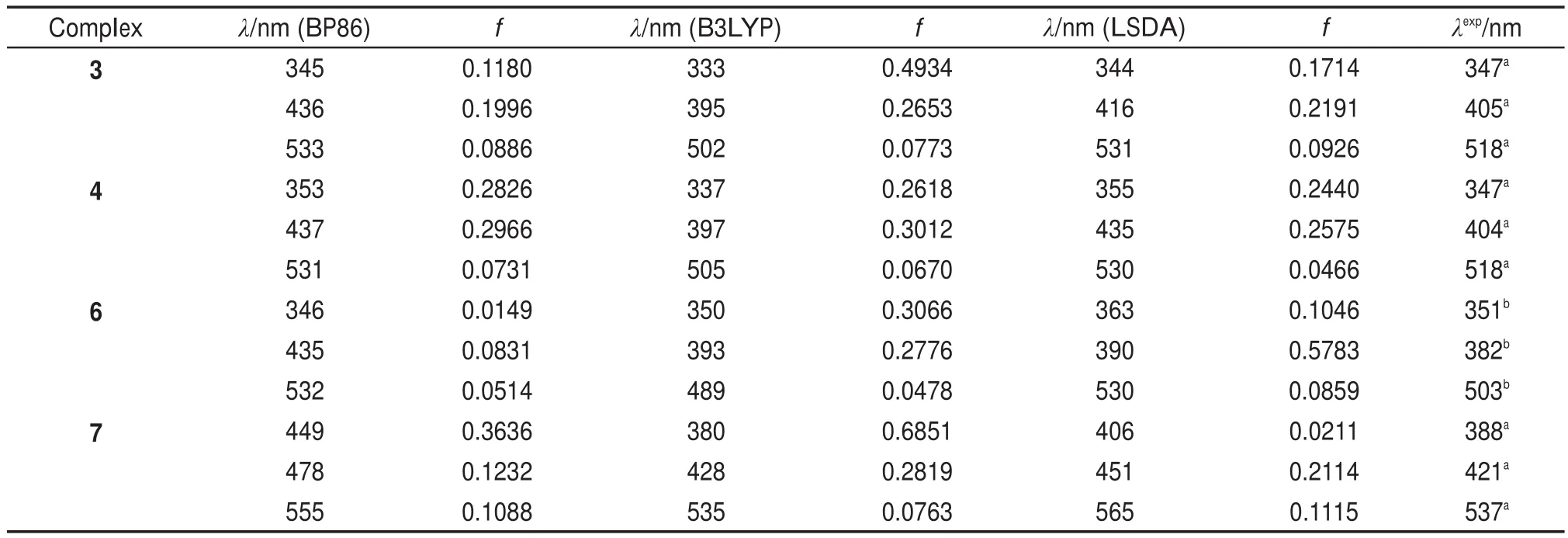
表3 配合物3、4、6和7的电子吸收光谱的实验值与相应的多种DFT计算值Table 3 Experimental and calculated electronic spectra of 3,4,6 and 7 by various DFT methods
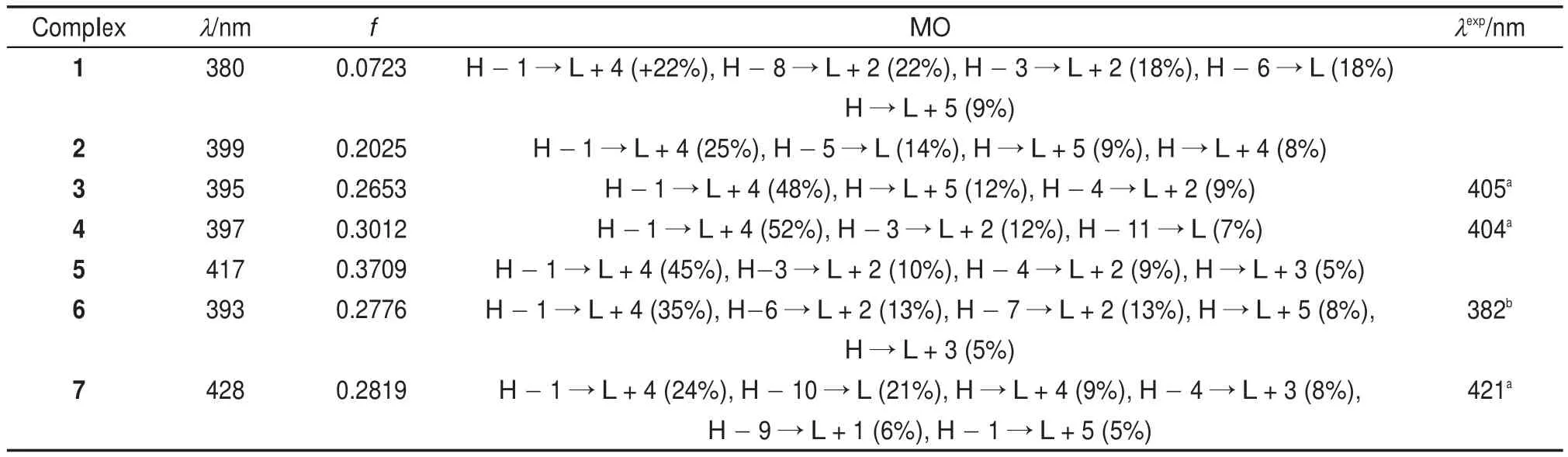
表4 配合物1-7的Soret吸收带的实验值和B3LYP方法的计算值Table 4 Experimental and calculated Soret band values of 1-7 complexes by B3LYPmethod
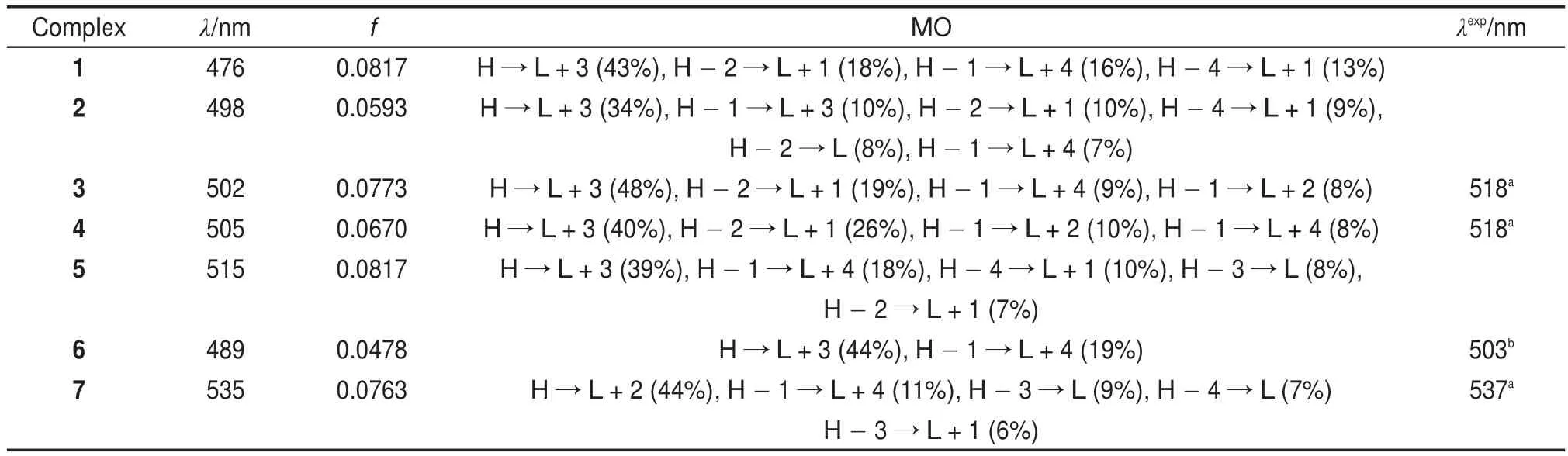
表5 配合物1-7的Q吸收带的实验值和B3LYP方法的计算值Table 5 Experimental and calculated Q bands value of 1-7 complexes by B3LYPmethod
3.4配合物的前线轨道分析

图3 配合物1-7的Soret带相关分子轨道能级图Fig.3 Molecular orbital energy levels of 1-7 complexes for Soret bands
配合物的光谱特征与前线分子轨道性质密切相关。为探讨电子跃迁实质,从前线轨道能级图(图 3)可以看出,HOMO轨道能量为6<7<4<5,表明随着β位取代基拉电子效应的逐渐减弱,HOMO轨道能级升高。β位无取代基时HOMO轨道能量顺序是4<3<2<1。化合物1-7的ΔEL-H分别为0.06933、0.07009、0.06929、0.06926、0.06672、0.06922和0.06730 e,其顺序从小到大为:4<3<1<2;5<7<4<6,与相应电子吸收光谱变化的情况不一致。尽管Liang等30报道在咔咯磷(V)氧化合物的光谱研究中,利用HOMO-LUMO轨道能隙的变化解释了Q带的红移现象。但是本文上述化合物的HOMO-LUMO轨道能隙与光谱红移的规律不明显(见图3和图4)。
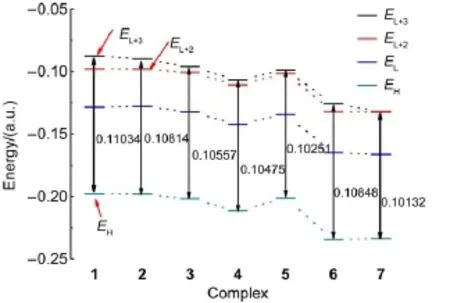
图4 配合物1-7的Q带相关分子轨道能级图Fig.4 Molecular orbital energy levels of 1-7 complexes for Q bands
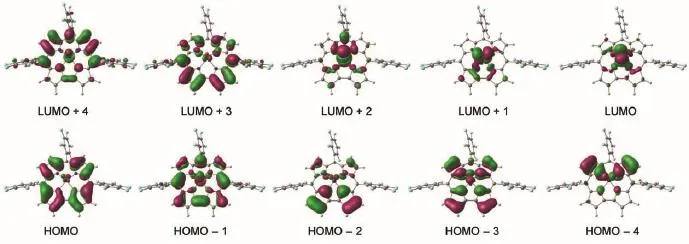
图5 配合物4的分子轨道图Fig.5 Molecular orbitals of complex 4
配合物1-7的前线轨道相似,现以配合物4的轨道图形(如图5)为例,它的HOMO与HOMO-1集中分布在咔咯骨架上,取代基、中位苯环与锰(V)氧基团上的分布非常少;HOMO基本分布在四个吡咯的碳原子上,中位碳原子上的分布较少。HOMO-1分布在中位碳原子和吡咯的氮原子上。从表4和表5中我们知道Soret带和Q带分别是HOMO-1到LUMO+4之间和HOMO到LUMO+ 3之间的跃迁(虽然配合物7的Q带主要由HOMO 到LUMO+2引起,但是LUMO+2轨道分布形状与其他化合物的LUMO+3是类似的,所以7个化合物的Q带跃迁轨道类型是相同的)。表4与图3和表5与图4中分别列出这两组轨道的能级差。其中,配合物7的两组轨道能级差最小。根据取代位置分两组体系讨论,其能级差ΔEL+4-H-1在中位和β位的取代的顺序从小到大分别为7<5<4<6;2< 4<3<1,而Soret带计算值从大到小的顺序(7> 5>4>6;2>4>3>1),说明能级差越小,红移的程度越大。同时Soret带计算结果与实验现象相符(配合物7的值明显大于3、4和6的值)。另外,考察Q带的情况,在跃迁的两个轨道之间,其能级差ΔEL+3-H的顺序从小到大分别是:7<5<4< 6;4<3<2<1,而Q带计算值从大到小的顺序(7>5>4>6;4>3>2>1),同样说明能级差越小,发生红移的程度越大。进一步观察上述轨道的分布情况,发现HOMO和HOMO-1反映咔咯环上的吡咯五元环间中位碳上的π成键轨道,而LUMO+3(对于化合物7来说是LUMO+2)和LUMO+4对应于π反键轨道。配合物5和7上的β取代基都是空间位阻较大的基团,导致咔咯环变形,进而削弱中位碳上的π键,致使相应ΔE能级差减小,最终引起对应的Soret带和Q带发生红移。
4 结论
对β位取代基修饰7个咔咯锰(V)氧配合物的电子吸收光谱进行理论计算。结果显示由取代基的空间效应导致的咔咯环扭曲是上述化合物电子吸收光谱Soret带和Q带红移的重要因素。咔咯环的扭曲会削弱中位碳上的π键轨道,导致Soret带和Q带的ΔE能隙相应减小,最终引起Soret带和Q带发生红移。
References
(1)Johnson,A.W.;Kay,I.T.J.Chem.Soc.1965,1620. doi:10.1039/JR9650001620
(2)Kadish,K.M.;Erben,C.;Ou,Z.;Adamian,V.A.;Will, S.;Vogel,E.J.Inorg.Chem.2000,39,3312.doi:10.1021/ ic991361m
(3)Gross,Z.;Galili,N.;Simkhovich,L.;Saltsman,I.;Botoshansky, M.;Bläser,D.;Boese,R.;Goldberg,I.J.Org.Lett.1999,1, 599.doi:10.1021/ol990739h
(4)Paolesse,R.;Mini,S.;Sagone,F.;Boschi,T.;Jaquinod,L.; Nurco,D.J.;Smith,K.M.Chem.Commun.1999,No.14,1307. doi:10.1039/A903247I
(5)Gryko,D.T.;Jadach,K.J.Org.Chem.2001,66,4267. doi:10.1021/jo010146w
(6)Aviv,I.;Gross,Z.Chem.Commun.2007,No.20,1987. doi:10.1039/B618482K
(7)Wasbotten,I.H.;Wondimagegn,T.;Ghosh,A.J.Am.Chem. Soc.2002,124,8104.doi:10.1021/ja0113697
(8)Thomas,K.E.;Wasbotten,I.H.;Ghosh,A.J.Inorg.Chem. 2008,47,10469.doi:10.1021/ic802317e
(9)Alemayehu,A.;Hansen,L.K.;Ghosh,A.J.Inorg.Chem.2010, 49,7608.doi:10.1021/ic1008736
(10)Alemayehu,A.;Conradie,J.;Ghosh,A.Eur.J.Inorg.Chem. 2011,2011,1857.doi:10.1002/ejic.201001026
(11)Ghosh,A.;Wondimagegn,T.;Parusel,A.B.J.J.Am.Chem. Soc.2000,122,5100.doi:10.1021/ja9943243
(12)Thomas K,E.;Conradie,J.;Hansen,L.K.;Ghosh,A.Eur.J. Inorg.Chem.2011,2011,1865.doi:10.1002/ejic.201100170
(13)Ghosh,A.;Steene,E.J.Biol.Inorg.Chem.2001,6,739. doi:10.1007/s007750100275
(14)Steene,E.;Dey,A.;Ghosh,A.J.Am.Chem.Soc.2003,125, 16300.doi:10.1021/ja021158h
(15)Sun,E.J.;Wang,D.;Cheng,X.L.;Shi,Y.H.;Shi,T.S.Chem. J.Chin.Univ.2007,28,1208.[孙二军,王栋,程秀利,师宇华,师同顺.高等学校化学学报,2007,28,1208.]
(16)Sun,E.J.;Cheng,X.L.;Wang,D.;Zhuang,C.F.;Xia,A.Q.; Shi,T.S.J.Coord.Chem.2009,62,1584.doi:10.1080/ 00958970802680674
(17)Yuan,W.;Ren,Q.J.;Sun,H.D.;Li,H.;Cheng,Y.X.;Ma,D. G.Chem.J.Chin.Univ.2014,35,1229.[袁伟,任清江,孙恒达,李慧,程延祥,马东阁.高等学校化学学报,2014,35, 1229.]
(18)Liu,H.Y.;Mahmood,M.H.;Qiu,S.X.;Chang,C.K.Coordin. Chem.Rev.2013,257,1306.doi:10.1016/j.ccr.2012.12.017
(19)He,J.;Xu,Z.G.;Zeng,Y.X.;Xu,X.;Yu,L.;Wang,Q.;Liu,H. Y.Acta Phys.-Chim.Sin.2012,28,1658.[何婧,徐志广,曾允秀,许旋,喻兰,王琦,刘海洋.物理化学学报,2012, 28,1658.]doi:10.3866/PKU.WHXB201205101
(20)Gong,L.Z.;Xu,Z.G.;Xu,X.;He,J.;Wang,Q.;Liu,H.Y.Acta Phys.-Chim.Sin.2014,30,265.[龚丽珍,徐志广,许旋,何婧,王琦,刘海洋.物理化学学报,2014,30,265.] doi:10.3866/PKU.WHXB201312181
(21)Kumar,A.;Goldberg,I.;Botoshansky,M.;Buchman,Y.;Gross, Z.J.Am.Chem.Soc.2010,132,15233.doi:10.1021/ja1050296 (22)Gross,Z.;Golubkov,G.;Simkhovich,L.J.Angew.Chem.Int. Edit.2000,39,4045.doi:10.1002/1521-3773(20001117)39:22< 4045::AID-ANIE4045>3.0.c0;2-p
(23)Liu,H.Y.;Lai,T.S.;Yeung,L.L.;Chang,C.K.Org.Lett. 2003,5,617.doi:10.1021/ol027111i
(24)Zhang,R.;Harischandra,D.N.;Newcomb,M.Chem.Eur.J. 2005,11,5713.doi:10.1002/chem.200500134
(25)Collman,J.P.;Zeng,L.;Decreau,R.A.Chem.Commun.2004, No.24,2974.doi:10.1039/B310763A
(26)Liu,H.Y.;Fei,Y.;Xie,Y.T.;Li,X.Y.;Chang,C.K.J.Am. Chem.Soc.2009,131,12890.doi:10.1021/ja905153r
(27)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, RevisionA.01;Gaussian Inc.:Pittsburgh,PA,2003.
(28)Gouterman,M.J.Mol.Spectrosc.1961,6,138.doi:10.1016/ 0022-2852(61)90236-3
(29)Vitasovic,M.;Gouterman,M.;Linschitz,H.J.Porphyr, Phthalocyanines 2001,5,191.doi:10.1002/jpp.309
(30)Liang,X.;Mack,J.;Zheng,L.M.;Shen,Z.;Kobayashi,N. Inorg.Chem.2014,53,2797.doi:10.1021/ic402347w
Effect of β-Substituents on the Electronic Absorption Spectra of Manganese(V)-oxo Corrole Complexes
CHEN Hua-Bin1ZHANG Xiao-Hui1GONG Li-Zhen1HE Jing1XU Xuan1XU Zhi-Guang1,*LIU Hai-Yang2,*
(1Key Laboratory of Theoretical Chemistry of Environment,Ministry of Education,School of Chemistry and Environment, South China Normal University;Guangzhou 510006,P.R.China;2Department of Chemistry,South China University of Technology,Guangzhou 510641,P.R.China)
Time-dependent density functional theory(TDDFT)calculations using the B3LYP method were used to investigate the effect of β-substituents on the electronic absorption spectra of a series of manganese(V)-oxo corrole complexes.The geometries,frontier molecular orbitals,and energies of the complexes were analyzed. The calculated results indicate that the steric effect of the β-substituents is the factor controlling the red-shift of the Soret bands and Q bands in these complexes.The sterically hindered β-substituents cause twisting of the corrole ring structure,which leads to reduced orbital energy gaps and a red-shift of the Soret bands and Q bands.
Manganese(V)-oxo corrole;Electronic absorption spectrum;TDDFT;Substituent
January 11,2016;Revised:April 20,2016;Published on Web:April 22,2016.
O641
10.3866/PKU.WHXB201604222
*Corresponding authors.XU Zhi-Guang,Email:chzgxu@scnu.edu.cn;Tel:+86-20-39310187.LIU Hai-Yang,Email:chhyliu@scut.edu.cn;
Tel:+86-20-22236805.
The project was supported by the National Natural Science Foundation of China(21171057,21275057,21371059)and Natural Science Foundation of Guangdong Province,China(S2012010008763).
国家自然科学基金(21171057,21275057,21371059)和广东省自然科学基金(S2012010008763)资助项目
©Editorial office ofActa Physico-Chimica Sinica
[Article]
猜你喜欢
中学生数理化(高中版.高考数学)(2022年2期)2022-04-26
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
新世纪智能(数学备考)(2021年4期)2021-08-06
化学与粘合(2020年4期)2020-09-11
中学生数理化(高中版.高考数学)(2020年3期)2020-05-25
生物工程学报(2020年1期)2020-03-12
高中生·天天向上(2018年1期)2018-04-14
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
