
沼液中溶解游离氨基酸的测定——柱前衍生-反相高效液相色谱法
2016-09-07 08:00:40李建华刘文静
中国环境科学 2016年8期
李建华,刘文静,李 宁
沼液中溶解游离氨基酸的测定——柱前衍生-反相高效液相色谱法
李建华,刘文静,李 宁*
(同济大学环境科学与工程学院,污染控制与资源化研究国家重点实验室,上海 200092)
针对沼液特点开发pH值调节(pH 10.2)旋蒸浓缩(去除氨氮和挥发性生物胺)联合3K Millipore超滤离心分离的预处理方法.并以邻苯二甲醛(OPA)和氯甲酸芴甲酯(FMOC-Cl)为柱前衍生化试剂,结合反向高效液相色谱和荧光检测分析,对沼液中溶解游离氨基酸(DFAA)进行了定性和定量考察.该方法保证了各氨基酸在一定浓度范围内呈现良好的线性关系(R2均大于0.99),标准样品中所选用的24种氨基酸,除谷氨酸回收率为70.5%外,其它氨基酸回收率为89%~115%,相对标准偏差为1.1%~5.0%.用所建立的方法对样品中溶解游离氨基酸的测定结果为:原料即剩余污泥中的溶解游离氨基酸量为2.0mol/L,5%和20%含固率反应器的沼液中的溶解游离氨基酸量分别为0.04mol/L和1.94mol/L,且种类不同,初步表明了不同厌氧消化反应系统中氨基酸的利用和产生机制的差异.
超滤;旋转蒸发;反相高效液相色谱;溶解游离氨基酸;沼液
近年来,剩余污泥的产生量伴随着城镇污水处理能力的不断提升而迅速攀升.厌氧消化作为一种在实现污泥稳定化同时还可产生绿色能源-沼气的技术,一直被认为是污泥处理处置的合适选择之一.而污泥经过厌氧消化后所产生的沼渣和沼液中各种有益和有害物质的含量将直接影响其土地利用的前景.氮是沼渣和沼液中重要营养元素之一,主要以氨态氮(NH3-N和NH4+-N)和有机氮(氨基酸、环己胺和氨基化合物等)的形态存在,此外还有少量的为NO3--N/NO2--N等[1].蛋白质作为污泥中有机氮的主要组成在厌氧消化过程中能被分解为溶解游离氨基酸并进一步转化为氨态氮(NH3-N,NH4+).由于氨基酸对植物生长特别是光合作用具有独特的促进作用进而提高作物品质,如增加维生素C和糖的含量[2],把握沼液和沼渣中氮基酸组分的存在形态及含量对指导污泥的综合利用尤为重要,而建立起快速、有效的沼液中氨基酸测定方法即为实现这一目标的关键环节.
在众多的氨基酸测定方法中, 沼液中总氨基酸量的测定主要通过茚三酮比色法[3],而沼液中的氨基酸组分的测定,主要以气相色谱法[4]和高效液相色谱法[5-6]为主.由于大多数氨基酸无紫外吸收和荧光发射特征,为提高分析检测灵敏度和分离选择特性,通常需将氨基酸进行柱前或柱后衍生化,并选用阳离子交换或反相液相色谱法对其进行分离并经紫外或荧光检测(OPA/FMOC-Cl/RT-HPLC)来实现各组分的测定[7-11].能使氨基酸产生荧光的衍生剂有邻苯二甲醛(OPA)、9-氯甲酸芴甲酯(FMOC-Cl)、丹酰氯(DANSYL-Cl)等.沼液中溶解游离氨基酸的含量较低,并且沼液中复杂的其它成分以及所包含的许多细小固体悬浮物会对衍生化过程产生干扰:如高浓度氨氮和挥发性胺容易与某些待测氨基酸形成荧光副产物,干扰氨基酸的分析与测定.孟庆国等[4]采用气相色谱法测定了沼液中的18种溶解游离氨基酸,然而,该研究中所采用的分析方法较为繁琐,且未涉及样本加标回收率、精密度等参数的进一步分析.氨基酸是一类包含至少一个羧基和一个氨基官能团的化合物(等电点2.8~10.8),它在不同的pH条件下,会以阳离子、阴离子和中性分子的形式存在.可针对氨基酸这一特点选用被广泛地应用于目标物质分离和富集[12-13]的固相萃取法(SPE)以快速、经济、灵活和高效的方式去除其他干扰物并对氨基酸进行富集.然而,对污泥中溶解游离氨基酸进行测定的研究较少,前处理部分样本上样条件及其洗脱条件等的信息也十分缺乏,限制了该方法在沼液中溶解游离氨基酸分析前处理中的应用.
本研究对比分析了SPE固相萃取在不同pH值条件下的处理效果,同时,针对氨基酸相对于其他干扰物分子量小(70~204)的特点,通过pH值调节结合旋蒸去除高浓度氨氮和挥发性胺的方式,并进一步采用3K Millipore超滤离心作为沼液中氨基酸分析前处理的重要手段,最终确立了pH值调节旋蒸联合超滤离心并OPA/FMOC-Cl- RT-HPLC分析这种简单、快速、重现性好、回收率高的污泥中溶解游离氨基酸的检测方法,实现了沼液中24种溶解游离氨基酸(天冬氨酸、丝氨酸、色氨酸、谷氨酸、甘氨酸、组氨酸、精氨酸、苏氨酸、丙氨酸、脯氨酸、半胱氨酸、酪氨酸、缬氨酸、蛋氨酸、异亮氨酸、亮氨酸、天门冬酰胺、谷氨酰胺、瓜氨酸、正缬氨酸、赖氨酸、羟脯氨酸、肌氨酸及苯丙氨酸)的同步分析.
1 材料与方法
1.1 仪器和试剂
Agilent-1260液相色谱仪,配有在线脱气机,四元梯度泵,标准自动进样器,荧光及紫外检测器(FLD和UV)及Agilent化学工作站.超滤离心管(3K,Millipore),SCX-SPE固相萃取柱购自上海安谱科学仪器有限公司.
17种氨基酸的混合标准包括:天冬氨酸、丝氨酸、色氨酸、谷氨酸、甘氨酸、组氨酸、精氨酸、苏氨酸、丙氨酸、脯氨酸、半胱氨酸、酪氨酸、缬氨酸、蛋氨酸、异亮氨酸(ILE)、亮氨酸及苯丙氨酸.补充氨基酸包括:天门冬酰胺、谷氨酰胺、瓜氨酸、正缬氨酸、赖氨酸、羟脯氨酸、肌氨酸.衍生化试剂为邻苯二甲醛(OPA,HPLC)、3-巯基丙酸(3-MPA,HPLC)和9-氯甲酸芴甲酯(FMOC-Cl,HPLC),均为Sigma (St Louis, MO, USA)产品.甲醇、乙腈为HPLC 级试剂(阿拉丁),实验用水是Milli-Q水( Millipore,USA),其他试剂均为优级纯.
1.2 试剂配制
氨基酸混合标准液的配制是将浓度为2.5nmol/μL的氨基酸标准溶液用0.1mol/L HCl配制成浓度分别为5、25、50、100、200、450pmol/μL的混合标准溶液.
OPA衍生化试剂的配制是将80mg OPA 溶解在7mL pH值为10.2的40mmol/L硼酸缓冲液中,加入125μL 3-巯基丙酸和1ml乙腈.该溶液避光陈化90min 以上,以降低试剂空白.配置好的OPA衍生化试剂保存在-20℃冰箱中,最长可使用1年.
FMOC-Cl衍生化试剂的配制是将50mg FMOC-Cl溶解在10ml乙腈溶液中.该溶液保存在-20℃冰箱中,最长可使用1年.
1.3 RT-HPLC色谱条件
色谱柱为Agilent Zorbox Eclipse AAA柱(3.5μm,4.6mm×150mm).流动相A为0.04mol/L的NaH2PO4(用10mol/L NaOH 将pH值调至7.8); B为超纯水:乙腈:甲醇以10:45:45(V /V/V)的比例配置的混合溶液.荧光检测波长的条件设置为:x= 340nm,m= 460nm, PMT=10; 15.25min更换波长为x= 266nm,m=305nm, PMT=9.流动相的流速为2mL/min,柱温设置为40 ℃.流动相洗脱梯度见表1.
1.4 样品前处理
使用0.22μm滤膜过滤沼液样品.过滤后的沼液用1mol/LNaOH溶液调节pH值至10.2,旋蒸浓缩至干后用0.1mol/L HCl重新溶解;SCX固相萃取实验用的样品在过滤后通过1mol/L HCl调节pH值至2.2待用.
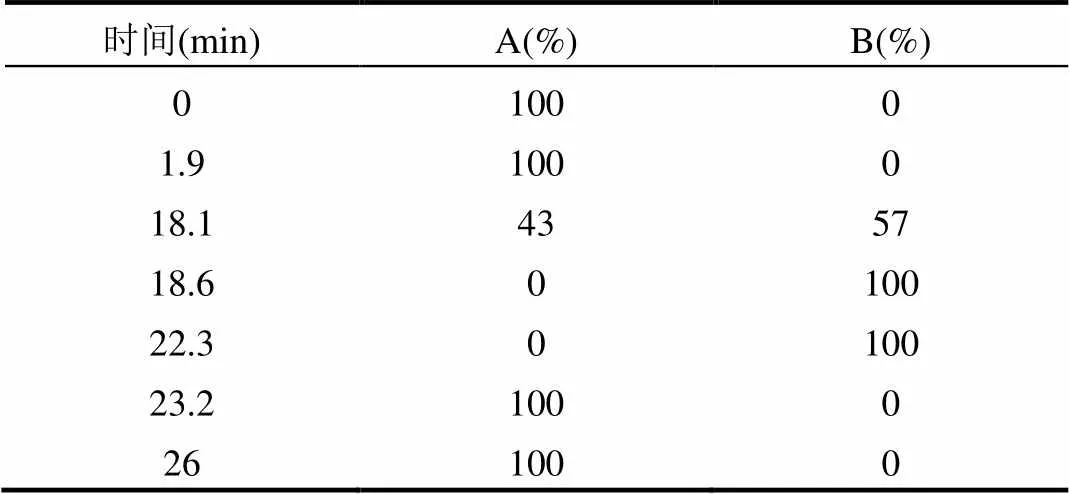
表1 流动相洗脱梯度 Table 1 Scheme of elution gradient for HPLC-FLD analysis
1.4.1 SCX固相萃取小柱回收率实验 实验中所用SPE固相萃取小柱经6mL甲醇和6mL水活化与平衡,控制流速小于3mL/min.取5mL沼液样品调节pH值至10.2并过SCX-SPE固相萃取小柱,控制流速小于1mL/min,此步骤不收集过滤液,然后用6mL 2%甲酸冲洗固相萃取小柱,最后先用5mL甲醇洗脱(此步骤亦不收集洗脱液),再用5%氨化甲醇洗脱,收集洗脱液.取洗脱液5ml于50mL茄型旋蒸瓶中,装上防爆瓶,于40℃水浴下负压旋转蒸发至干.然后用5mL 0.1mol/L HCl重新溶解.
1.4.2 3K Millipore超滤膜前处理 将上述pH值调节至10.2的沼液5mL转移至50mL茄型旋蒸瓶中,装上防爆瓶,于40℃水浴下负压旋转蒸发至干,然后用5mL 0.1mol/LHCl重新溶解.涡旋30s以混合均匀,取上述液体200μL,采用离心的方式通过3K Millipore超滤离心管,收集滤液.
1.5 氨基酸的衍生化
依据文献中所报道的方法对OPA的衍生化步骤进行了部分调整[14].首先将0.5μL的样品混合0.5μL的OPA和0.5μL的FMOC,而后用32μL的混合溶液(100mL流动相A加400μL的磷酸) 进行稀释,将所得溶液通过HPLC进行分析.整个过程耗时近2min,该混合过程可通过安捷伦HPLC自动进样程序完成以严格控制各反应时间.
1.6 回收率试验
将100μmol/L的氨基酸标样溶解于空白水体中,进行上述两种方法的回收率试验.另取同样浓度的氨基酸标样加于样品中测定样本加标回收率.
2 结果和讨论
2.1 前处理方法的优化
采用保留时间法对样品中氨基酸进行定性分析.即在相同的色谱分析条件下,样品中氨基酸的色谱峰与氨基酸标准的色谱峰保留时间相同或相近,则认为样品中含有此类氨基酸.在每次衍生化反应之前,采用外标工作曲线法对样品中溶解游离氨基酸的浓度进行定量分析.
2.1.1 SCX-SPE条件的优化及加标回收率实验 由于氨基酸在不同的pH条件下会以阳离子、阴离子和中性分子的形式存在.文献中一般选用(强)阳离子基质的SPE柱提取血浆中的氨基酸[15]和果汁、果酱中的氨基酸[16-17].但不同文献中所选用的样品pH值不同,且缺少对样品pH值影响的深入讨论.为此,本研究中采用同一沼液样品,比较不同pH值对其中氨基酸提取效果的影响.如图1所示,在pH值为2.2时,样本中氨基酸的保留效果最好.
如表1所示,即便在最优pH值(pH 2.2)的条件下,采用SCX-SPE法得到的空白样品各氨基酸加标回收率差异较大(0%~96%),相对标准偏差为0.6%~8%.对于天门冬酰胺、谷氨酰胺、苏氨酸、瓜氨酸、酪氨酸、缬氨酸、蛋氨酸、正缬氨酸、色氨酸、苯丙氨酸、异亮氨酸和亮氨酸,该方法具有较高的回收率(大于80%),可用于这些氨基酸的定性和定量分析;而对天冬氨酸、谷氨酸、丝氨酸、组氨酸、甘氨酸、精氨酸、丙氨酸、赖氨酸、羟脯氨酸、肌氨酸、脯氨酸和半胱氨酸的回收率较低(小于80%,0~76%不等).因而,采用SCX-SPE法前处理对部分氨基酸的选择性较强,对其他氨基酸进行定量分析时会存在较大的误差.而采用3K Millipore超滤离心管前处理方法得到空白水体中各氨基酸加标回收率为97.9%~110.1%,相对标准偏差为0.2%~2.7%(表2),24种氨基酸可同步定性和定量分析.
同时,考虑到沼液中干扰组分对回收率的影响,本研究中也设计了样品加标回收率的实验.通过SCX-SPE固相萃取小柱前处理的沼液样本中各氨基酸的加标回收率为0%~101.2%,相对标准偏差为2.7%~25.6%,各氨基酸的回收率差别较大,对超过半数的氨基酸进行定量分析时会存在较大的误差.而通过3K超滤离心管前处理的样本各种氨基酸的回收率为70.5%~115.1%,相对标准偏差为1.1%~5.0%(表2).除谷氨酸之外的其他23种氨基酸的同步定性和定量分析得以实现.
2.1.2 氨氮浓度对于色谱分析结果的影响 沼液内氨氮浓度可高达3000mg/L,相对于低含量的溶解游离氨基酸而言,高浓度氨氮在极大程度上会影响溶解游离氨基酸的分析.这是由于沼液中浓度较高的氨和挥发性胺在衍生化过程中易形成荧光副产物,与某些氨基酸同时洗脱出来,影响溶解游离氨基酸的定性定量分析.研究表明,衍生化反应前,沼液中的氨和挥发性胺的浓度需低于10-5mol/L,才能解决氨基酸液相色谱图模糊和峰重叠的问题[7].当采用SCX-SPE前处理方法进行分析时,由于有5%氨化甲醇洗脱及旋蒸浓缩至干的步骤,氨氮得以去除,避免氨氮及挥发性胺的影响;而当采用3K Millipore超滤膜前处理的方法时却易受到高浓度氨氮的影响,故需要通过调节pH和旋蒸的步骤进一步去除氨氮.

表2 不同前处理方法的各氨基酸的样本加标回收率和精密度(n=3) Table 2 Spike recovery of 24amino acids in slurry samples with different pretreatments (n=3)
如图2(a)所示,未经氨氮去除前处理的沼液中部分氨基酸的各色谱峰无法正常分离,导致无法对这些氨基酸进行定量;图2(b)中经过pH的调节和后续的旋蒸浓缩前处理后,高浓度氨氮的干扰得以去除,色谱图效果明显好于图2(a).
2.1.3 两种不同前处理方法对比 沼液样品经超滤与SCX-SPE两种方法前处理后的对比图如图3所示.经SCX前处理后,沼液中部分氨基酸流失无法定量(图3(a));经超滤并去除氨氮的前处理后,沼液中氨基酸损失少(图3(b)),色谱图效果明显好于图3(a).
2.2 氨基酸测定的线性范围、回归方程、相关系数、精密度
24种浓度为100pmol/μL 的氨基酸混合标准溶液色谱分离图如图4所示.从图中可以看出,在上述衍生化和色谱条件下,实现了24种氨基酸的彻底分离.将所配制的5种不同浓度的氨基酸混合标准液,按1.3的色谱条件进行测定,以峰面积为横坐标(X),以氨基酸标样浓度为纵坐标(Y),进行线性回归得到回归方程(表3).结果表明,各氨基酸的相关系数在0.991~0.9998,即在5~450pmol/μL 的线性范围内氨基酸峰面积与其质量浓度呈良好的线性关系.同时,混合氨基酸标准品溶液重复进样7次,24种氨基酸的出峰时间及氨基酸浓度RSD均在2.2%以内(表4),说明本分析中仪器的精密度较好.
2.3 方法的应用
将上述建立的基于超滤前处理联合柱前衍生-HPLC测定沼液中氨基酸的分析方法用于不同反应器沼液中溶解游离氨基酸的含量的研究.如表5所示,作为进料的剩余污泥中溶解游离氨基酸的含量为2.0mol/L.进料含固率为5%和20%的反应器中溶解游离氨基酸的种类不同,含量分别为0.04mol/L和1.94mol/L,氨基酸含量和种类与含固率之间并无明显关系.该结果初步表明,不同厌氧消化过程对氨基酸的利用和产生机理不同.因而,还需要在所开发的方法的基础上进一步研究厌氧消化过程中蛋白质的降解和转化的机理.

表3 标准样品的线性回归分析结果 Table 3 Results of linear regression analysis of calibration data

猜你喜欢
化工管理(2022年14期)2022-12-02 11:43:52
云南化工(2021年8期)2021-12-21 06:37:36
科学导报·学术(2020年29期)2020-10-21 10:49:43
环境科技(2016年6期)2016-11-10 05:14:06
农业知识(2016年26期)2016-03-27 22:44:29
上海蔬菜(2016年5期)2016-02-28 13:18:05
环境科技(2015年3期)2015-11-08 12:08:34
实用手外科杂志(2015年4期)2015-08-27 01:54:06
现代检验医学杂志(2015年4期)2015-02-06 02:02:19
农村农业农民·B版(2014年10期)2014-11-07 21:02:15
