
两种含吡唑基1,3,4-噁二唑衍生物的合成及光谱性质*
2016-09-01 09:34许文君游金宗胡飞飞杨春霞边佳莹齐德强
广州化工 2016年2期
许文君,游金宗,胡飞飞,杨春霞,边佳莹,黄 娟,齐德强
(浙江外国语学院科学技术学院,浙江 杭州 310012)
两种含吡唑基1,3,4-噁二唑衍生物的合成及光谱性质*
许文君,游金宗,胡飞飞,杨春霞,边佳莹,黄娟,齐德强
(浙江外国语学院科学技术学院,浙江杭州310012)
以苯乙酮为起始原料,与草酸二乙酯、水合肼反应得到5-苯基-1H-吡唑-3-甲酸乙酯,再通过亲核取代反应、酯的肼解反应和合环反应,得到5-(1-苯甲基-3-苯基-1H-吡唑-5-基)-2-硫基-1,3,4-噁二唑,在碳酸钾催化下,进一步与对甲基溴化苄发生亲核取代反应得到5-(1-苯甲基-3-苯基-1H-吡唑-5-基)-2-(4-甲基苯甲基-2-硫基)-1,3,4-噁二唑。所有中间体及目标化合物的结构均经核磁共振、红外光谱、元素分析表征,并对目标化合物的紫外吸收光谱和荧光光谱性质进行了初步研究。
吡唑;噁二唑;合成;光谱
杂环化合物具有良好的荧光性能和广泛的生物活性,逐渐成为化学、医药、农药领域的研究热点。吡唑类衍生物具有诸如抗菌、消炎、降低血糖、抗癌等生物活性,许多吡唑衍生物已被开发为杀菌剂、杀虫剂、杀螨剂、除草剂及医用药物[1-3]。噁二唑类化合物不仅具有除草、杀菌、杀虫、抗癌等生物活性,还因具有较好的荧光性能而同样备受到人们的关注,一些噁二唑衍生物可作为荧光剂、闪烁剂和激光材料,有些衍生物还可以作为感光高分子应用于电发光仪器[4-6]。此外,利用它们的结构特点,对噁二唑骨架进行改造[7],有望合成出功能多样的杂环衍生物。
近年来,人们报道了众多吡唑环、噁二唑环衍生物在光学及生物活性方面的应用研究[8-11]。尽管如此,通过官能团修饰,将吡唑环引入到1,3,4-噁二唑母体,并进行适当取代,合成吡唑连噁二唑硫醚(醇)类的化合物鲜有报道[12-13]。基于此,本文以苯乙酮为起始原料,经过多步反应,合成了两种含吡唑基的1,3,4-噁二唑衍生物,5-(1-苯甲基-3-苯基-1H-吡唑-5-基)-2-硫基-1,3,4-噁二唑(4)和5-(1-苯甲基-3-苯基-1H-吡唑-5-基)-2-(4-甲基苯甲基-2-硫基)-1,3,4-噁二唑(5),化合物的结构经过核磁共振(1H NMR)、红外光谱(IR)、元素分析(EA)表征确证,研究了化合物的紫外吸收和荧光性质。
1 实验部分
1.1仪器与试剂
试剂:苯乙酮;草酸二乙酯;水合肼(80%);氯化苄;对甲基溴化苄;二硫化碳;碳酸钾;氢氧化钾;二甲基亚砜;乙酸;乙腈;甲醇;乙醇;乙酸乙酯;石油醚(60~90 ℃)等均为市售分析纯。
分析仪器:Nicolet iS10傅立叶变换红外光谱仪,美国Thermo Fisher公司;AVANCE 300 MHz核磁共振谱仪。瑞士Bruker公司;PE-240C型元素分析仪,美国Perkin-Elmer公司;UV-2550紫外-可见分光光度计,日本Shimadzu公司;FP-6200荧光光谱仪,日本JASCO公司;WRR熔点仪,上海精密科学仪器有限公司;RE52-99旋转蒸发器,上海亚荣生化仪器厂;78HW-1数显恒温磁力搅拌器,杭州仪表电机有限公司。
1.2实验方法
化合物1~5的合成路线如图1所示。
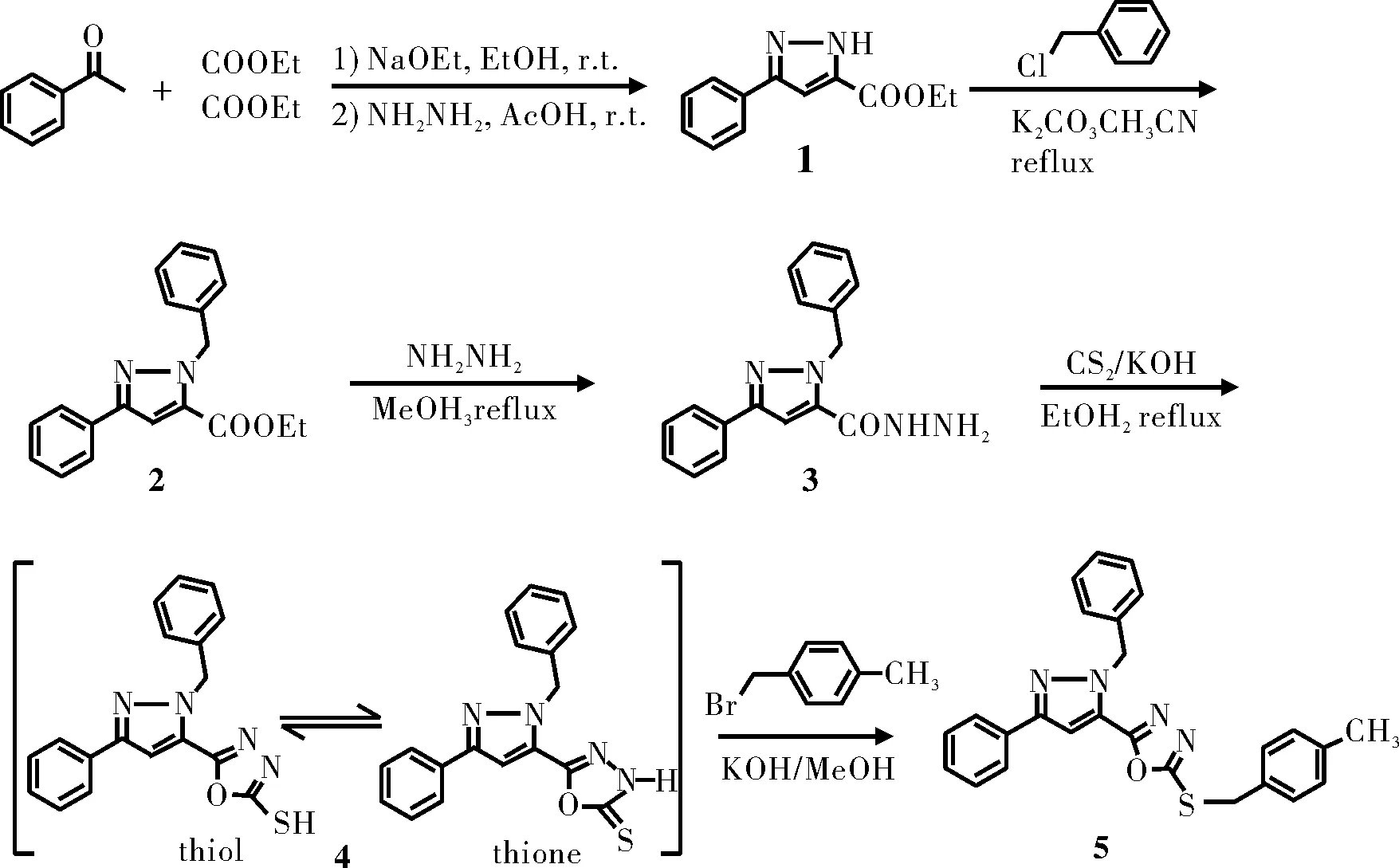
图1 化合物的合成路线
1.2.13-苯基-1H-吡唑-5-甲酸乙酯(1)
向250 mL三口烧瓶中加入金属钠(2.07 g,0.09 mol)和无水乙醇(100 mL),搅拌至完全溶解,滴加草酸二乙酯(10.35 g,0.07 mol),搅拌均匀后,开始滴加苯乙酮(8.8 mL,0.07 mol),控制滴加时间1 h,滴加完毕后,室温反应3 h,向反应瓶中加入冰醋酸(10.3 mL,0.15 mol),搅拌1 h后,滴加80%水合肼(6 mL,0.1 mol),1.5 h滴完,继续反应10 h,减压蒸去大部分溶剂,抽滤,滤饼用水、石油醚洗涤,粗产品用乙酸乙酯进行重结晶,得白色固体,产率74%,熔点:138~140 ℃。IR(KBr,ν,cm-1):3460(br),3111(w),2906(w),1727(s),1615(m),1468(m),1419(m),1242(w),1195(m),1139(m),1024(m),762(s)。1H NMR(300 MHz,CDCl3,25 ℃,TMS,ppm):7.74~7.69(m,2H,ArH),7.34~7.20 (m,3H,ArH),7.06~7.02(m,1H,Pyrazole,=C-H),4.37~4.28(q,2H,CH2),1.37~1.28(t,3H,CH3)。
1.2.21-苯甲基-3-苯基-1H-吡唑-5-甲酸乙酯(2)
向50 mL圆底烧瓶中加入化合物1(1.08 g,5.24 mmol),碳酸钾(0.71 g,5.14 mmol),乙腈(25 mL),搅拌15 min后加入氯化苄(0.79 g,6.24 mmol)。加热回流,用TLC监测至反应终点,减压蒸去溶剂,加入二氯甲烷,过滤,减压浓缩除去溶剂,所得粗产品用V(乙酸乙酯):V(石油醚)=1:5作洗脱剂,硅胶柱色谱分离,得到白色晶体,产率66.1%,熔点92~93 ℃(文献值[8]:92~94 ℃)。IR(KBr,ν,cm-1):3460(br),3142(w),2984(w),1720(s),1453(m),1325(m),1261(s),1096(m),763(s),728(m),697(m)。1H NMR(300 MHz,CDCl3,25 ℃,TMS,ppm):7.83~7.81(m,2H,ArH),7.81~7.81(d,2H,ArH),7.32~7.28(t,5H,ArH),7.27~7.23(t,1H,ArH),7.15(s,1H,Pyrazole,=C-H),5.80(s,2H,NCH2),4.33~4.28(m,2H,CH2),1.36~1.33(t,3H,CH3)。
1.2.31-苯甲基-3-苯基-1H-吡唑-5-碳酰肼(3)
向化合物2(3.36 g,10.9 mmol)的甲醇(5 mL)溶液中加入80%水合肼(12 mL),加热回流反应至原料完全消耗,减压蒸去大部分溶剂,过滤,得粗产品,用少量乙醇重结晶得白色絮状固体,产率89.8%,熔点:131~132 ℃(文献值[13]132~133 ℃)。IR(KBr,ν,cm-1):3452(br),3302(w),3277(w),3197(w),2982(w),1680(s),1613(s),1544(m),1447(s),1328(m),1208(m),1103(w),772(s),723(m),696(s)。1H NMR(300 MHz,CDCl3,25 ℃,TMS,ppm):7.77~7.75(d,2H,ArH),7.40~7.36(m,2H,ArH),7.32~7.21(m,6H,ArH),6.79(s,1H,Pyrazole,=C-H),5.77(s,2H,NCH2),3.81(bs,1H,NH)。
1.2.45-(1-苯甲基-3-苯基-1H-吡唑-5-基)-2-硫基-1,3,4-噁二唑(4)
向50 mL圆底烧瓶中加入化合物3(0.50 g,1.71 mmol),乙醇(20 mL),KOH(0.25 g,4.46 mmol),搅至澄清,再缓慢滴加二硫化碳( 0.39 g,5.13 mmol),回流反应7 h,减压蒸发除去大部分溶剂,将残余物慢慢倒入冰水中,用乙酸调节pH=5~6,析出沉淀,过滤,水洗,得到类白色粗产品,再经乙醇重结晶得到白色固体,产率85.1%,熔点214~215 ℃。元素分析,(C18H14N4OS,%):实测值为C 64.73,H 4.32,N 16.68;理论值为C 64.65,H 4.22,N 16.75。IR (KBr,cm-1,v):3096(br),3001(w),2941(w),2811(w),2769(w),1639(s),1507(s),1483(s),1455(s),1329(w),1305(s),1183(m),1055(s),963(s),761(m),730(s),683(m)。1H NMR(300 MHz,CDCl3,25 ℃,TMS,ppm):10.52(s,1H,SH),7.84~7.82(d,2H,ArH),7.44~7.21(m,9H,ArH),5.75(s,2H,NCH2)。
1.2.55-(1-苯甲基-3-苯基-1H-吡唑-5-基)-2-(4-甲基苯甲基-2-硫基)-1,3,4-噁二唑(5)
向50 mL圆底烧瓶中加入化合物4(1.0 g,3 mmol),碳酸钾(0.557 g,4 mmol),乙腈(25 mL),充分搅拌后,慢慢加入氯化苄(0.45 g,3.6 mmol),加热回流,用TLC监测至反应终点,减压蒸发除去溶剂,慢慢加入冰水中,充分搅拌,抽滤,依次用水,少量乙醇洗涤,得白色粉末,产率73.1%,熔点123~124 ℃。元素分析(C26H22N4OS,%):实测值为C 71.38,H 5.04,N 12.72;理论值为C 71.21,H 5.06,N 12.78。IR(KBr,ν,cm-1):3064(w),3028(w),2987(w),2947(w),2922(w),1617(m),1468(s),1417(w),1289(w),1161(m),1057(s),810(m),763(s),721(s),689(s)。1H NMR(300 MHz, CDCl3,25 ℃,TMS,ppm):7.86~7.84(d,2H,ArH),7.36~7.24(m,10H,ArH),7.16(d,2H,ArH),7.10(s,1H,Pyrazole,=C-H);5.91(s,2H,NCH2),4.48(s,2H,SCH2),2.34(s,3H,CH3)。
2 结果与讨论
2.1红外光谱分析
采用KBr压片法,在Nicolet iS10傅立叶变换红外光谱仪上测定了化合物的红外谱图,化合物4和5的红外吸收光谱分别如图2(a)和(b)所示,在3096和3028 cm-1附近的吸收峰分别对应化合物4和5上的不饱和C-H键伸缩振动,在3001~2769 cm-1和2987~2922 cm-1处的弱吸收峰对应4和5的为饱和C-H键伸缩振动。文献[14]报道1,3,4-噁二唑-2-硫醇存在“硫醇-硫酮”互变异构现象,化合物4的红外谱图中没有出现S-H特征吸收峰(2600~2550 cm-1),并且在1305 cm-1处出现C=S键特征吸收峰,说明在固体状态下,化合物4的主要存在形式为硫酮异构体,N-H的吸收峰则与水峰重叠。
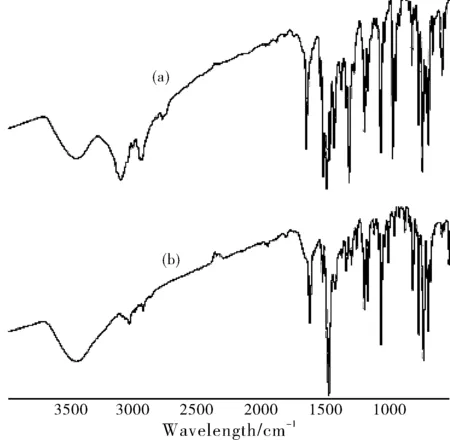
图2 化合物4(a)和5(b)的红外光谱
2.2核磁共振分析
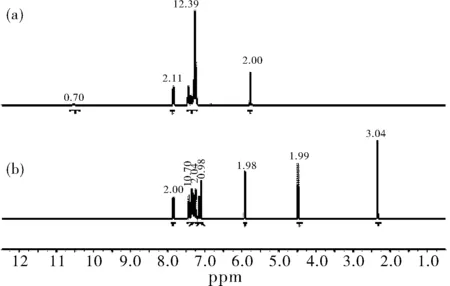
图3 化合物4(a)和5(b)的核磁谱图
以DCCl3为溶剂,以四甲基硅烷(TMS)为内标,在核磁共振仪上测定了化合物的核磁共振谱图。化合物4的核磁谱图如图3(a)所示,在化学位移为10.52 ppm处出现宽的单峰,对应S-H,而N-H的特征峰[2]没有出现,证明化合物4在DCCl3中的主要存在形式为硫醇异构体。在7.84~7.82 ppm处的双重峰对应Ar-H,7.44~7.21 ppm的多重峰对应Ar-H和吡唑环上的=C-H,在5.75 ppm处的单峰则对应亚甲基-CH2,化合物5的核磁谱图如图3(b)所示,该化合物的核磁共振共有七组信号,化学位移为7.86~7.84 ppm的二重峰对应于与甲基相连的苯环上的Ar-H,7.36~7.24 ppm的多重峰对应于苯环上的Ar-H,7.16 ppm的二重峰对应于与甲基相连的苯环上的Ar-H,7.10 ppm 的单峰对应于吡唑环上的=C-H,5.91 ppm的单峰对应于与吡唑环相连的亚甲基上的N-CH2,4.48 ppm的单峰对应于与硫相连的亚甲基上的S-CH2,2.34 ppm的单峰对应于甲基上的CH3。
2.3紫外吸收光谱分析
将化合物4和5配制成浓度为1.0×10-5mol/L的DMSO溶液,用UV-2550紫外-可见分光光度计对其进行紫外光谱扫描,如图4所示,化合物4特征吸收峰为300 nm,摩尔消光系数为9.35×104L/(mol·cm),化合物5特征吸收峰为271 nm,摩尔消光系数为7.52×104L/(mol·cm)。与化合物4相比,化合物5的紫外吸收发生蓝移(29 nm),摩尔消光系数也显著降低,这是由于化合物4的氢原子被对甲苯甲基取代之后,使得“苯环-吡唑-噁二唑”三环的刚性变差,共轭性降低,导致化合物5吸光度明显减弱,吸收峰位置向短波长方向移动,产生蓝移现象。

图4 化合物4和5的紫外光谱图
2.4荧光光谱分析
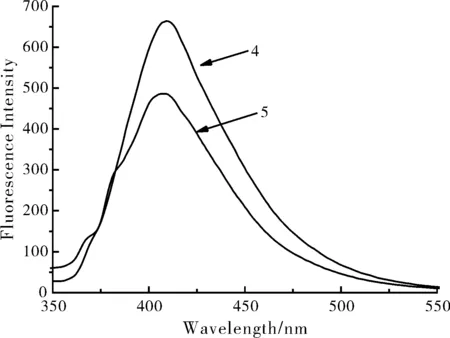
图5 化合物4和5的荧光光谱图(激发波长:334 nm)
将化合物4和5配制成浓度为1.0×10-5mol/L的DMSO溶液,以334 nm为激发波长,在350~550 nm范围内对其进行荧光光谱扫描,所得荧光光谱如图5所示。由图可知,化合物4的最大发射波长为409 nm,化合物5的最大发射波长为408 nm。两个化合物的最大发射波长没有发生明显改变,但与化合物4相比,化合物5的荧光强度明显降低,这是由于化合物4的氢原子被对甲苯甲基取代之后,使得“苯环-吡唑-噁二唑”三环的刚性变差,荧光发生部分淬灭,导致荧光强度降低。
3 结 论
以苯乙酮为原料,通过五步反应,生成了两种含吡唑基1,3,4-噁二唑衍生物。通过核磁共振、红外吸收、元素分析表征了它们的结构,并初步研究了它们的紫外吸收光谱和荧光光谱性质,为进一步设计合成具有良好荧光性能及生物活性的新型杂环类化合物奠定了基础。
[1]PATE M V, BELL R, MAJEST S, et al.Synthesis of 4,5-diaryl-1H-pyrazole-3-ol derivatives as potential COX-2 inhibitors[J].J Org Chem, 2004, 69(21): 7058-7065.
[2]PUTHIYAPURAYIL P, POOJARY B, CHKKANA C, et al.Design, synthesis and biological evaluation of a novel series of 1,3,4-oxadiazole bearing N-methyl-4-(trifluoromethyl) phenyl pyrazole moiety as cytotoxic agents[J].Eur J Med Chem, 2012, 53: 203-210.
[3]刘志昌,王应红,张元勤,等.姜黄素-N-取代吡唑类衍生物合成及抑菌活性[J].有机化学,2012,32:1487-1492.
[4]王凯凯,张齐贤,李亚丰,等.含偶氮苯基元对称双二唑衍生物的合成及其发光性能[J].应用化学,2013,30(1):48-53.
[5]FENG LH, CHEN Z B.Light emitting conjugated molecule containing 1,3,4-oxadiazole,carbazole and naphthalene units[J].Spectrochica Acta Part A, 2006, 63(12): 15-20.
[6]BRAIN C T, PAUL J M, LOONG Y, et al.Novel procedure for the synthesis of 1,3,4-oxadiazoles from 1,2-diacylhydrazines using polymer supported Burgess reagent under microwave conditions[J].Tetrahedron Lett, 1999, 40(16): 3275-3278.
[7]宋庆宝,徐丽娟,马淳.1,3,4-噁二唑类化合物合成及应用的研究新进展[J].浙江化工,2009,40(8):17-23.
[8]丁晓玲,孟宁,夏永,等.1-芳甲基-3-芳基吡唑-5-羧酸乙酯衍生物的合成与生物活性评价[J].有机化学,2007,27(12):1542-1546.
[9]BHAT B A, DHARK L, PURI S C, et al. Synthesis and biological evaluation of chalcones and their derived pyrazoles as potential cytotoxic agents[J]. Bioorg Med Chem Lett, 2005,15: 3177-3180.
[10]CHENG D M, MA F Y, LIU X Y. Pure red emission of dye-doped organic molecules from microcavity organic light emitting diode[J]. Opt Laser Technol, 2007, 39(4):720-723.
[11]BONDOCK S, ADEL S, ETMAN H A, et al. Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles[J]. EJMed Chem, 2011, 48(26):192-199.
[12]SCHÜRER S C, BROWN S J, GONZALEZ-CABRERA P J, et al. Ligand-binding pocket shape differences between sphingosine 1-phosphate (S1P) receptors S1P1and S1P3 determine efficiency of chemical probe identification by ultrahigh-throughput screening[J]. ACS Chem Biol, 2008, 3(8): 486-498.
[13]XIA Y, DONG Z W, ZHAO B X, et al. Synthesis and structure-activity relationships of novel 1-arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazidederivatives as potential agents against A549 lung cancer cells[J]. Bioorg Med Chem, 2011, 111(11): 6984-7034.
[14]MANSOUR A K, E I D M M, KHALIL N S A M. Synthesis and reactions of some new heterocyclic carbohydrazides and related compounds as potential anticancer agents[J]. Molecules, 2003, 8(10): 744-755.
Synthesis and Spectrum Properties of Two Pyrazole-based 1,3,4-Oxadiazole Derivatives*
XUWen-jun,YOUJin-zong,HUFei-fei,YANGChun-xia,BIANJia-ying,HUANGJuan,QIDe-qiang
(School of Science and Technology, Zhejiang International Studies University, Zhejiang Hangzhou 310012, China)
Ethyl 3-phenyl-1H-pyrazole-5-carboxylate was obtained by the reaction of acetophenone with diethyl oxalate and hydrazine hydrate. 5-(1-benzyl-3-phenyl-1H-pyrazol-5-yl)-1,3,4-oxadiazole-2-thiol was synthesized by reactions of nucleophilic substitution, hydrazinolysis and cyclization, which further reacted with 1-(bromomethyl)-4-methylbenzene in acetone in the presence of potassium carbonate to afford 2-(1-benzyl-3-phenyl-1H-pyrazol-5-yl)-5-(4-methylbenzylthio)-1,3,4-oxadiazole. The structures of all intermediates and products were confirmed by1H NMR, IR and elemental analysis. In additions, the UV-vis and fluorescence spectrum of two title compounds were investigated.
pyrazole; oxadiazole; synthesis; spectrum
国家级大学生创新创业训练计划项目(201414275006)。
许文君(1994-),女,本科生,主要从事有机功能材料的合成。
齐德强(1982-),男,实验师,主要从事有机功能材料的合成。
O621.3
A
1001-9677(2016)02-0039-04
猜你喜欢
高中数理化(2022年2期)2022-02-22
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
含能材料(2020年7期)2020-07-11
农药科学与管理(2019年8期)2019-11-23
世界农药(2019年3期)2019-09-10
山西青年(2017年2期)2017-01-11
中学生数理化·高二版(2016年3期)2016-12-26
天然产物研究与开发(2016年11期)2016-06-15
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
