
鉴别猪伪狂犬病病毒野毒与疫苗毒双重PCR检测方法的建立
2016-08-30 01:08卢婷婷李存法陈忠杰连瑞丽
中国兽医杂志 2016年7期
赵 丽,卢婷婷,李存法,陈忠杰,连瑞丽
(河南牧业经济学院,河南 郑州 450011)
鉴别猪伪狂犬病病毒野毒与疫苗毒双重PCR检测方法的建立
赵丽,卢婷婷,李存法,陈忠杰,连瑞丽
(河南牧业经济学院,河南 郑州 450011)
依据基因库中的猪伪狂犬病病毒(PRV)基因序列,分别设计了gE和gH两对引物,以PRV闽A株、Bartha (gE-)株和Norden(Tk-)株为模板,筛选最佳反应条件,建立了检测猪伪狂犬病病毒野毒株与疫苗毒株的鉴别PCR方法。该方法能从PRV闽A株、Norden(Tk-)株中扩增出一条355 bp的条带,但Bartha(gE-)株没有扩增出该目的条带。对正常细胞、猪细小病毒(PPV)、猪圆环病毒(PCV)进行检测,结果均为阴性,没有出现交叉反应。在对单项PCR反应条件(引物浓度、Mg2+浓度、退火温度等)优化的基础上,建立了鉴别猪伪狂犬病病毒野毒与疫苗毒的双重PCR检测方法,并分别用双重PCR和单项PCR检测15份临床病料,两者符合率为97.5%,表明该双重PCR检测方法有较高的敏感度,可以用于临床病料的检测。
双重PCR;伪狂犬病病毒;野毒和疫苗毒;鉴别诊断
伪狂犬病病毒(Pseudorabies virus,PRV)是多种家畜和野生动物均可感染的一种疱疹病毒,常引起母猪出现繁殖障碍及初生仔猪大批死亡,成年猪则长期带毒排毒,给养猪业造成极大的经济损失。近几年养殖场猪伪狂犬病又有上升趋势,控制和消灭伪狂犬病所面临的主要困难是确定伪狂犬病病毒的潜伏感染、野毒感染或疫苗感染,猪在初次感染康复后往往长期带毒,当机体受到内外因素刺激时,病毒被激活并持续排毒[1-4],引起疫病的扩散。疫苗免疫接种和疾病的早期鉴别诊断是有效防制和根除伪狂犬病的根本措施。目前常用的方法就是PCR扩增,快速、特异,因此,有必要建立一种快速、早期并能同时确定是疫苗株感染还是野毒株感染的双重PCR检测方法[5-7]。
gH基因是伪狂犬病病毒中较保守和病毒复制所必需的基因;gE基因是伪狂犬病病毒的毒力基因也是常用的基因缺失疫苗的缺失基因。以gH、gE基因序列作为靶序列建立的PCR技术,能够区分基因缺失疫苗接种与野毒感染[6]。因此本研究根据猪伪狂犬病病毒gH、gE基因序列设计两对引物,建立了鉴别猪伪狂犬病病毒野毒与疫苗毒的双重PCR方法。
1 材料与方法
1.1毒株与菌种PRV闽A(Fa)株、Norden(Tk-)株和Bartha(gE-)株、猪圆环病毒(PCV)、猪细小病毒(PPV),由河南省动物性食品安全重点实验室保存;PRVg基因E缺失疫苗,购自武汉科前生物制品有限公司。工程菌JM109感受态细胞为宝生物工程(大连)有限公司产品;病料采自河南周边猪场。
1.2质粒工具酶及其他化学试剂EX TaqDNA聚合酶为北京康为生物工程有限公司产品;异硫氰酸胍(GuSCN)、重蒸饱和酚、酚∶氯仿∶异戊醇(25∶24∶1)、2.5 mmol/L dNTP;2 000 bp DNA lad⁃der、200 bp DNA ladder、DNA凝胶回收试剂盒、溴化已锭(EB)、溴酚兰等均为北京康为世纪生物科技有限公司产品;琼脂糖(Agarose):Promega公司产品。
1.3主要仪器CO2培养箱、PCR仪、紫外微量分光光度计,购自美国Thermo Forma公司;DYY-III-8B稳压稳流型电泳仪、电泳槽:北京六一仪器厂;超速冷冻离心机:SIGMA公司;紫外凝胶成像系统:Alpha Innotech公司。
1.4方法
1.4.1引物的设计与合成利用DNAStar(Ver⁃sion5.0)软件进行同源性分析,应用Primer Pre⁃mier5.0软件设计出扩增gH基因355 bp和gE基因139 bp的几对特异引物
gH:上游引物(P1):5′-ACGGAAGTTATA⁃AGGATGA-3′;
下游引物(P2):5′-TGTCTCCGAAGAAATAA⁃GA-3′;
gE:上游引物(P3):5′-ACGGAAGTTATA⁃AGGATGA-3′;
下游引物(P4):5′-TGTCTCCGAAGAAATAA⁃GA-3′。
两对引物均由北京三博远志生物技术有限责任公司合成。
1.4.2PRV病毒培养及模板DNA的制备用PK15细胞培养伪狂犬病病毒株,在细胞病变达75%以上时收获细胞,反复冻融3次,置-20℃保存备用。病毒DNA及临床样品模板DNA制备参照[8]。
1.4.3伪狂犬病病毒gH、gE基因单一PCR检测方法的建立及优化
1.4.3.1伪狂犬病病毒gH、gE基因PCR检测方法的建立按如下体系进行PCR扩增:

反应条件94℃变性3 min后,进人循环94℃30 s,56℃(gH)和54℃(gE)30 s,72℃30 s,25个循环后,72℃延伸5 min。扩增产物用1.0%琼脂糖凝胶(含0.5 μg/mL EB)进行电泳检测。
1.4.3.2gH、gE基因单一PCR反应条件的优化
对gH、gE基因单一PCR反应参数包括引物、Taq酶、dNTPs、Mg2+浓度及退火温度、时间、循环次数等进行优化,建立两种基因的单联PCR最佳反应体系。
1.4.4单一PCR检测方法的特异性试验分别提取PRV Fa株、Bartha(gE-)株和Norden(Tk-)株、PRV gE基因缺失疫苗和PPV、PCV等病毒DNA,分别进行PCR扩增,并设立阴性对照,以检测该单一PCR检测方法的特异性。
1.4.5单一PCR检测方法的敏感性测定提取PRV Fa毒株DNA,测定其含量后,将其核酸依次作10倍的梯度稀释,采用己优化好的PCR反应体系进行PCR扩增,以检测其敏感性。
1.4.6gH、gE基因双重PCR检测方法的建立在50 μL体系中加入以下成份:


瞬时离心后于PCR仪上94℃预变性5 min后,94℃30 s;56℃30 s;72℃1 min,30个循环,最后72℃再延伸5 min。同时设立无模板的阴性对照。反应结束后,取5 μL PCR产物用1.5%琼脂糖凝胶(含0.5 μg/mL EB)进行电泳检测PCR结果。
1.4.6.1双重PCR反应体系的优化(1)双重PCR的引物浓度的优化:gH、gE基因分别以50 pmol/μL的引物浓度0.2 μL、0.4 μL、0.6 μL、1 μL进行双重PCR反应,以选取两种病毒的最佳引物浓度;(2)双重PCR的退火温度的优化:gH、gE基因分别以52℃、54℃、56℃、58℃的退火温度进行双重PCR反应,以确定最佳的退火温度;(3)双重PCR检测方法的特异性试验:分别提取PRV Fa株、Bartha(gE-)株和Norden(Tk-)株的DNA,加入己优化好的双重PCR体系中进行扩增,并以PPV、PCV及正常PK15细胞作为阴性对照,以检测该双重PCR检测方法的特异性;(4)双重PCR检测方法的敏感性试验:参照[8],超速离心纯化PRV Fa株病毒及gH、gE基因的DNA,10倍的梯度稀释并测定其病毒滴度,采用己优化好的双重PCR体系进行扩增,以检测其敏感性。
1.4.6.2对病料进行复合PCR检测对采自河南不同地区的猪伪狂犬病疑似病料进行双重PCR检测。
2 结果与分析
2.1单一PCR优化检测方法的扩增结果提取PRV Fa的标准强毒株DNA,进行单一的PCR优化扩增反应。其电泳结果显示,PCR扩增出一条约355 bp和139 bp目的DNA片段,与预期大小相当(图1、图2),PCR产物纯化回收后测序表明,所扩增序与参照的PRV核酸序列同源性为100%,证明PCR产物为预期扩增的目的DNA片段。
2.2单一PCR检测方法的特异性分析用gH、gE基因特异性的引物分别对PRV Fa株、Bartha株、Norden株进行扩增,并将猪常见的繁殖障碍性疾病病原PPV、PCV和正常PK15细胞作为阴性对照,进行PCR扩增。结果如图3、图4。
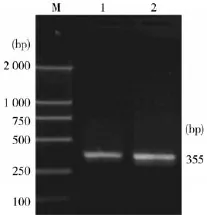
图1 PRVgH基因PCR扩增结果
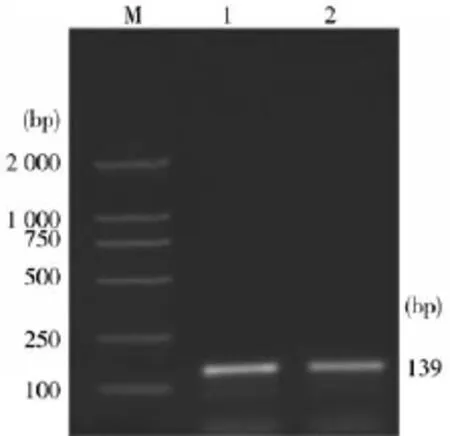
图2 PRVgE基因PCR扩增结果

图3 PRVgH PCR特异性测定

图4 PRVgE PCR特异性测定
PRV的所有毒株均可以扩增出355 bp的片段,PRV Fa株和Norden株可以扩增出139 bp的片段,而阴性对照样品均未能扩增出任何条带。
2.3单一PCR检测方法的敏感性测定结果将提取的PRV Fa株的DNA按照病毒滴度作梯度稀释,以优化好的单一PCR反应体系进行PCR扩增,其结果如图5。

图5 PCR检测PRVgH模板DNA的敏感性
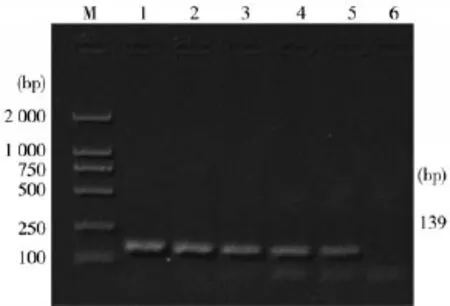
图6 PCR检测PRVgE模板DNA的敏感性
gH基因的单一PCR方法最低可检测到大约为10 TCID50/100 μL的病毒DNA;gE基因的单一PCR方法最低可检测到大约为9.5 TCID50/100 μL的病毒DNA。
2.4双重PCR检测方法的扩增结果提取PRV Fa株、Bartha(gE-)株的DNA后,进行双重PCR扩增反应,其结果如图7。

图7 双重PCR反应扩增的结果
从图7可看出,双重PCR可扩增出PRV gH基因355 bp和gE基因139 bp的条带,证明该双重PCR可同时扩增PRV gH基因和gE基因从而鉴别野毒与疫苗毒感染。
2.5双重PCR反应体系的优化结果
2.5.1引物浓度的优化将PRV gH、gE基因分别以50 pmol/μL,30 pmol/μL,20 pmol/μL,10 pmol/ μL的引物浓度进行双重PCR反应,结果显示,gH基因的最佳引物浓度为20 pmol/μL,gE基因的最佳引物浓度为20 pmol/μL。
2.5.2双重PCR退火温度的优化将PRV gH、gE基因分别以52℃、54℃、56℃、58℃的退火温度进行双重PCR反应,在退火温度为56℃时扩增结果均清晰无杂带。
2.6双重PCR方法特异性试验结果分别应用gH、gE基因的特异性引物,以优化好的双重PCR体系,对PRV Fa、Bartha株、Norden株、gE基因缺失疫苗株以及对照样品进行双重PCR扩增,其扩增结果如图8。
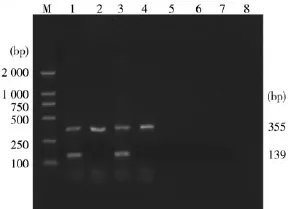
图8 双重PCR方法特异性结果
由图8可以看出,PRV Fa株、Norden株均出现355 bp和139 bp特异性条带,而Bartha株和gE基因缺失疫苗株只出现355 bp条带,对照样品则呈阴性,说明了该双重PCR反应良好的特异性。
2.7双重PCR方法敏感性测定结果将提取PRV gH、gE基因的DNA按照其病毒滴度进行系列稀释,利用己优化好的双重PCR反应体系进行双重PCR扩增,其结果如图9。该二联PCR最低能检测到10-4稀释PRV即gH 100 TCID50/100 μL,gE 95 TCID50/100 μL的核酸模板。
2.8重复性试验结果重复检测PRV的样品3次,结果均一致。
2.9双重PCR病料检测结果对15份来自河南周边地区PRV疑似病料检测,其中有5份双重PCR检测gH基因为阳性;有3份双重PCR检测gH、gE基因同时为阳性;其余病料复合PCR检测全为阴性,在同样条件处理下的健康猪材料为阴性,结果与单项PCR检测结果符合率达97.5%。

图9 双重PCR方法敏感性测定结果
3 结论与讨论
3.1PCR技术具有快速和灵敏度高、特异性强等优点,但常规的PCR技术一次检测不能区分出疫苗毒还是野毒感染。本研究根据伪狂犬病病毒PRV基因缺失疫苗缺失了gE基因和病毒必需基因gH的特点,按照PRV gE和gH的序列设计两对引物,建立了特异性强、敏感性高、稳定性好,能区分gE基因缺失疫苗毒和野毒的PCR鉴别诊断方法。该方法同时能用于潜伏感染病毒的检测,可区分开野毒潜伏感染与疫苗毒潜伏感染。复合PCR反应是一种特殊的PCR形式,当其反应体系中同时含有两对或两对以上引物和模板时,反应总是有利于片段较小的一方,即短片段优先扩增的原理[2]。但是本试验gE虽然是短片段但并没有优先扩增,这可能与模板量差异以及毒株差异有密切关系。gE基因G+C%含量高达74.4%,开始时用一般的10×Buffer扩增不出特异性条带,后来换成2×GC BufferII后得到很好扩增。本试验所扩增的PRV gH、gE片段为355 bp和139 bp,两者相差不大,在选择反应条件时,尽量选择有利于gE基因的扩增条件,以达到最佳的复合PCR扩增效果。
3.2本试验在单项PCR条件的基础上作出了复合PCR反应条件的选择。在引物浓度的选择上,考虑到gE基因难扩增,所以选择两者单项PCR扩增均较好的引物浓度。在温度循环参数中,由于两种PCR反应的变性和延伸条件相同,故没作改变。复性温度gH基因在54℃、56℃扩增效果较好,而gE基因在54℃、56℃、58℃均能得到很好扩增,故复合PCR的复性温度选择在56℃。从PRV双重PCR的敏感性测定结果表明,在双重PCR反应中,只要引物设计合理,反应条件选择得当,双重PCR扩增和常规PCR技术一样,都具有极高的敏感性,能检出痕量目的DNA片段。
3.3对15份临床病例检测结果表明,5份gH基因检测阳性为使用了伪狂犬病病毒gE基因缺失疫苗,3份gH、gE基因同时为阳性为伪狂犬病病毒野毒感染,由此可见,应用PRV的双重PCR技术可以直接对临床样品进行扩增,可区分野毒和疫苗毒,大大提高了PRV的诊断速度,这对PRV的早期诊断及区分疫苗毒和野毒临床的快速鉴别诊断具有很高的实用价值。
[1]Yoon H A,Eo S K,Aleyas A G,et al.Investigation of pseudora⁃bies virus latency in nervous tissues of seropositive pigs exposed to field strain[J].Vet Med Sci,2006,68(2):143-148.
[2]Yoon H A,Eo S K,Aleyas A G,et al.Molecular survey of latent pseudorabies virus infection in nervous tissues of slaughtered pigs by nested and real-time PCR[J].Microbiol,2005,43(5):430-436.
[3]Tanaka S,Mannen K.Effect of mild stress in mice latently infect⁃ed pseudorabies virus[J].Exp Anim,2003,52(5):383-386.
[4]Jin L,Schnitzlein W M,Scherba G.Identification of the pseudorabies virus promoter required for latency-associated transcript gene ex⁃pression in the natural host[J].Virology,2000,74(14):6333-6338.
[5]Palladino S,Kay I,Fonte R.Use of real-time PCR and the light cycler system for the rapid detection of Pneumocystis carina in re⁃spiratory specimens[J].Diagn Microbiol infect Dis,2001,39(4):233-236.
[6]周斌,苏鑫铭,张素芳,等.PCR快速检测伪狂犬病病毒野毒感染[J].中国病毒学,2004,19:612-615.
[7]赵丽,崔保安,方忠意,等.PCR检测猪伪狂犬病病毒方法的研究[J].中国预防兽医学报,2007,29(2):142 146.
S852.65
A
0529-6005(2016)07-0012-04
2015-11-16
河南省重点科技攻关项目(152102110092)
赵丽(1973-),女,副教授,博士生,研究方向为分子病毒学与免疫学,E-mail:zhaolisq@163.com
猜你喜欢
江苏安全生产(2022年9期)2022-11-02
动漫星空(兴趣百科)(2020年3期)2020-03-24
中国动物传染病学报(2020年1期)2020-02-09
中国外汇(2019年7期)2019-07-13
智富时代(2019年2期)2019-04-18
智富时代(2019年2期)2019-04-18
猪业科学(2018年4期)2018-05-19
中国兽医杂志(2017年11期)2018-01-11
中华实验和临床病毒学杂志(2017年5期)2017-01-13
科学启蒙(2016年7期)2016-08-06
