
婴幼儿神经系统遗传代谢病的脑MRI研究
2016-08-22 10:56程爱兰曹雯君汪登斌
国际医学放射学杂志 2016年4期
程爱兰 冯 赟 侯 亮 曹雯君 汪登斌 金 彪
婴幼儿神经系统遗传代谢病的脑MRI研究
程爱兰冯赟侯亮曹雯君汪登斌金彪
目的探讨婴幼儿神经系统遗传代谢病的脑MRI表现。方法收集2014年1月—12月我院诊治的16例遗传代谢病病人临床及影像资料,并结合临床特点及实验室检查分析该组病人的脑MRI表现。结果16例遗传代谢病病人中包括有机酸血症12例(甲基丙二酸血症8例,枫糖尿病4例),尿素循环障碍3例,异染性脑白质营养不良1例。12例有机酸血症中,7例基底节区信号异常,主要表现在T1FLAIR上呈低信号,T2FLAIR上呈稍高信号,DWI上呈明显高信号;4例脑干及2例小脑半球在DWI上呈明显高信号,2例脑萎缩,1例胼胝体在DWI上呈高信号,1例双侧半卵圆中心在T1FLAIR上呈低信号、T2FLAIR高信号、DWI高信号,1例脑室扩大。3例尿素循环障碍病人中2例脑室扩大,1例大脑皮质在DWI上呈弥漫性高信号。1例异染性脑白质营养不良表现为两侧侧脑室周围白质、半卵圆中心T2FLAIR高信号。结论婴幼儿神经系统遗传代谢病脑MRI表现缺乏特异性,根据疾病类型不同MRI表现各异,其中有机酸血症患儿以基底节区受累为主。
遗传代谢病;脑;磁共振成像;甲基丙二酸血症;枫糖尿病
Int J Med Radiol,2016,39(4):339-343
遗传代谢病(inborn metabolic diseases,IMD)为一组特定的脑疾病,通常由基因突变导致的一个或多个新陈代谢途径发生生化改变而引起。无论是正常生化物质缺乏,还是对脑实质具有毒性作用的异常生化物质聚集,均可导致脑损伤并引起相应的临床症状。遗传代谢病的发生率大约为40/10万,80%病例以神经系统症状首发[1]。该组疾病临床表现缺乏特异性,易误诊、漏诊。因此本文回顾性分析16例IMD病人临床及脑MRI资料,以提高对本病的认识。
1 资料与方法
1.1临床资料收集2014年1月—12月间于上海交通大学医学院附属新华医院诊治的16例IMD病人临床及影像资料,其中男10例,女6例,确诊年龄为3 d~3岁,平均(11.9±11.0)个月。其中有机酸血症12例,包括甲基丙二酸血症(methylmalonic acidemia,MMA)8例,枫糖尿病 (maple syrup urine disease,MSUD)4例,尿素循环障碍(urea cycle disorders,UCD)3例,异染性脑白质营养不良(metachromatic leukodystrophy,MLD)1例。纳入标准:所有患儿均经气相色谱-质谱法(GC/MS)以及基因检测确诊,且均有完整脑MRI影像资料。排除标准:所有患儿均经气相色谱-质谱法确诊,且缺少基因检测或脑MRI影像资料其中一项;有维生素B12、叶酸缺乏等继发因素的病例。本研究获得上海交通大学医学院附属新华医院伦理委员会批准,所有患儿家长均签署知情同意书。
1.2检查方法
1.2.1血液生化检测及基因检测生化检测应用API 4000串联质谱仪 (美国生物应用系统公司)进行血或尿有机酸分析。应用血滤纸片过滤患儿外周血,加甲醇萃取后离心吹干,上机进样检测。基因检测通过采集患儿外周血2 mL,分离白细胞,利用试剂盒提取DNA,在ABI3700测序仪上正反双向测序。
1.2.2影像检查采用GE Signa 1.5 T twinspeed超导MR设备,8通道头颅线圈,受试者头部采用海绵垫固定。患儿口服10%水合氯醛镇静,剂量为0.5 mL/kg,待其入睡后进行扫描。选择层厚5 mm,层间距1 mm,FOV24 cm×24 cm。行横断面T1液体衰减反转恢复(T1-fluid attenuated inversion-recovery,T1FLAIR)序列(TR 2200 ms,TE 24ms)、T2LAIR(TR 7500 ms,TE 150 ms)扫描,以及矢状面及横断面T2WI (TR 2000 ms,TE 90 ms),横断面DWI(TR 10 000 ms,TE 70 ms,b=0和1000 s/mm2)扫描。
1.3生化、基因检测分析及影像分析血液生化检测结果中,若患儿尿甲基丙二酸、甲基枸橼酸异常增高,血丙酰肉碱、丙酰肉碱与乙酰肉碱比值不同程度增高,诊断为甲基丙二酸血症;分析显示亮氨酸、缬氨酸明显异常增高则诊断为枫糖尿病;若血浆瓜氨酸明显降低,尿乳清酸增高则诊断为尿素循环障碍;若白细胞溶酶体学分析显示芳基硫酯酶A活性明显降低则诊断为异染性脑白质营养不良。应用聚合酶链反应和直接测序法进行基因突变检测分析。MRI影像资料均由放射科2名经验丰富的医生独立单盲进行观察、分析,分析病灶的部位、各序列的信号特点,当2名医生意见不同时通过讨论达成一致结论。
2 结果
2.1血液生化及基因检测结果8例MMA病人GC/MS显示丙酰肉碱[(11.72±7.74)μmol/L,中位数为9.04 μmol/L;正常范围0.5~4.0 μmol/L]、丙酰肉碱与乙酰肉碱比值[0.72±0.29,中位数为0.75;正常范围0.05~0.25]、甲基枸橼酸[(15.07±11.85)mmol/mmolCr,中位数为11.52 mmol/mmolCr;正常范围0.0~1.1 mmol/mmolCr]、甲基丙二酸[(412.31±364.57)mmol/ mmolCr,中位数为310.59mmol/mmolCr;正常范围0.0~1.1 mmol/mmolCr]浓度不同程度增高,基因突变类型均为MUT基因突变。4例MSUD病人GC/MS显示亮氨酸、缬氨酸浓度明显增高,亮氨酸为(1 633.98± 886.29)μmol/L(中位数1 480.44 μmol/L,正常范围50.0~250.0 μmol/L),缬氨酸为(421.42±50.82)μmol/L(中位数为431.55 μmol/L,正常范围80.0~300.0 μmol/L);其中3例基因突变类型为BCKDHB,1例为BCKDHA。3例UCD中1例为瓜氨酸血症,基因检测显示ASS1基因突变;2例为鸟氨酸氨甲酰转移酶缺陷症(ornithine transcarbamylase deficiency,OTCD),其血浆瓜氨酸明显降低,分别为0.3 mg/L、0.1 mg/L[正常范围(3.0±1.0)mg/L],尿乳清酸均>10 mmol/ mmolCr(正常范围<10mmol/mmolCr)。1例MLD病人白细胞溶酶体学分析显示芳基硫酯酶A活性为5 nmol/(h·mg)[正常范围38.9~98.3 nmol/(h·mg)]。
2.2影像表现12例有机酸血症病人中,7例基底节区信号异常,主要表现在T1FLAIR上呈低信号,T2FLAIR上呈稍高信号,DWI上呈明显高信号 (图1);4例脑干及2例小脑半球在DWI上呈明显高信号(图2),2例脑萎缩,1例胼胝体在DWI上呈高信号,1例双侧半卵圆中心在T1FLAIR上呈低信号、T2FLAIR高信号、DWI高信号,1例脑室扩大。3例UCD病人中2例脑室扩大,1例大脑皮质在DWI上呈弥漫性高信号(图3)。1例MLD表现为两侧侧脑室周围白质、半卵圆中心T2FLAIR高信号,T2FLAIR上侧脑室周围、半卵圆中心白质呈条纹状交错排列(图4)。
3 讨论
遗传代谢病种类繁多,临床表现复杂,缺乏特异的临床表现,多表现为喂养困难、癫、抽搐、共济失调或脑标志区发育延迟。遗传代谢病的分类目前主要依据病人的临床症状、细胞特征性组织化学染色、生化代谢过程中破坏的细胞器以及潜在的基因突变部位和类型。目前尚缺乏根据影像特征进行分类的标准。了解不同疾病的影像特点并结合其临床表现有助于缩小鉴别诊断范围。

图1 患儿女,2岁。A横断面DWI示双侧苍白球呈对称性明显高信号,B T2FLAIR示双侧苍白球呈稍高信号,C T1FLAIR上双侧苍白球呈稍低信号。

图2 患儿男,42 d。A、B、C均为横断面DWI。A双侧小脑半球呈对称性高信号,脑桥呈高信号;B小脑蚓部、中脑呈高信号;C双侧背侧丘脑、双侧苍白球、双侧内囊后肢均呈高信号。
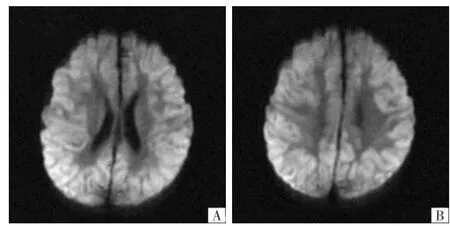
图3 患儿女,2岁。A、B均为横断面DWI,示大脑皮质呈弥漫性高信号,皮质下白质未见明显异常。

图4 患儿女,2岁。A、B均为横断面T2WI。T2WI示双侧额叶、双侧侧脑室三角区(A)、半卵圆中心呈高信号(B),白质内可见稍低信号条纹征。
本组病例中最多见的是有机酸血症,该类疾病是由于氨基酸、脂肪和糖代谢异常导致中间代谢产物——有机酸增加,从而引起一系列病理生理改变和临床症状的一组疾病。本研究中有机酸血症主要包括8例MMA,4例MSUD。MMA是支链有机酸血症中较常见的类型,主要由于甲基丙二酰辅酶A变位酶或其辅酶腺苷钴胺素缺陷,导致甲基丙二酰辅酶A生成琥珀酰辅酶A途径障碍,从而导致甲基丙二酸、丙酸和甲基枸橼酸等有机酸在体内大量蓄积,造成多脏器损伤,尤其是中枢神经系统损伤[2]。刘等[3]总结了56例MMA病人的影像特点,认为MMA最常见的征象是大脑和小脑脑沟增深,皮质萎缩,侧脑室扩张,与文献[4-5]报道一致。本研究报道的8例MMA病人中仅出现2例脑萎缩和1例脑室扩张(3/8例,37.5%)。一般早期病人以脑室扩张常见,发展到疾病晚期脑白质体积缩小直至消失,大脑呈萎缩样改变。这8例病人中最常见的MR征象是基底节区信号异常,其中有4例(50%)病变位于基底节区苍白球,MRI表现为T1WI低信号、T2WI高信号,DWI上明显高信号。有文献报道基底节区是MMA病人最常受累的部位,这主要因为MMA病人可能存在细胞色素C氧化酶和琥珀酸脱氢酶的活性降低,导致能量需求较高的基底节区更容易累及,毒性有机酸代谢物的蓄积也是苍白球损害的原因之一[6]。其中另1例半卵圆中心T2WI高信号,表明髓鞘化异常。MMA病人髓鞘化异常常位于侧脑室体部旁白质、双侧侧脑室前后角以及半卵圆中心。髓鞘化不良与代谢异常导致的脑内S-腺苷甲硫氨酸缺乏有关[7],如果不及时诊断治疗,病变发展到晚期这些异常髓鞘化的脑区白质体积减少,致使患儿预后较差。刘等[3]研究提出幕上白质水肿也是MMA的常见征象,但本组8例病人均未见明显的幕上脑白质水肿,可能是由于本组病例均为单纯型MMA。因为有文献报道MMA合并高胱氨酸血症病人幕上脑白质水肿较常见,且该类病人脑白质变性的主要病理改变是血管异常,是以血管损伤为主[7]。
MSUD是有机酸血症中较少见的类型,该病是由于基因缺陷导致支链氨基酸代谢异常,在新生儿中的发病率为1/12万~1/50万[8]。目前该病分为5型:经典型、中间型、间歇型、硫胺(维生素B1)有效型、二氢硫辛酰胺酰基脱氢酶(E3)缺乏型[9]。新生儿期发病者常为经典型,也最为严重。本研究中4例均为新生儿期发病,临床表现为嗜睡、反复抽搐、喂养困难,符合经典型。MSUD有特殊的MRI表现,即在急性失代偿期颅脑MRI表现出弥漫性水肿,包括侧脑室周围区域、脑干背侧及小脑深部白质[10]。近年来的一些研究发现在DWI上可观察到另一种类型的水肿,即MSUD水肿[11],这种特殊类型的水肿是由于扩散受限和扩散加快同时存在造成的,因其分布的部位具有特征性而得名,这些部位与足月新生儿髓鞘化的部位分布相一致,包括小脑深部白质、背侧脑干、大脑脚、内囊后支、半卵圆中心中部白质[12],并且已有研究证实通过电子显微镜可以观察到髓鞘内水肿[13]。王等[14]报道1例MSUD病人同时存在2种水肿,即MSUD水肿和弥漫性水肿。Righini等[15]研究认为DWI上高信号的髓鞘区域属于髓鞘内水肿,而DWI上低信号的非髓鞘区域是由于扩散加快,属于血管间质水肿。本组4例MSUD病人主要受累部位位于基底节区及脑干,在DWI上表现为明显高信号,与文献[10-11,14]报道一致。
本组12例有机酸血症病人均进行了DWI检查,其中7例基底节区出现明显高信号,T1WI、T2WI亦出现异常信号,T1WI上呈低信号,T2WI上呈稍高信号,但DWI显示病灶更加清晰,因此认为DWI对发现早期基底节区病变具有重要作用。
尽管基底节区信号异常是有机酸血症病人较特异的表现,但该征象发生率并不是很高[16]。因此,当脑内基底节区信号没有异常而出现其他部位信号异常时,需要与本组病例中的以下代谢性疾病鉴别。
UCD是一组由于先天酶缺陷引发的以高血氨为特征的代谢性疾病。尿素循环中任何一种酶的缺陷即可发生UCD,其中以OTCD最多见[17]。本组3例UCD病人中1例瓜氨酸血症,2例OTCD。罗等[18]报道1例OTCD病人脑MRI特点,发现该病人双侧基底节及丘脑信号呈短T1、短T2改变。与Takanashi等[19]报道的3例新生儿期发病的UCD影像改变一致。但Takanashi等研究进一步发现岛叶的白质在T2WI上呈高信号改变,而罗等报道病例中并未见到这一征象,考虑与疾病病情严重程度不同有关。本组3例UCD病人中2例脑室扩大,1例大脑皮质在DWI上呈弥漫性高信号,均与文献[18-19]报道不同。UCD中不同类型病人脑MRI表现存在明显差异,可能与生化机制及疾病过程有关。UCD导致神经系统受累的机制尚不完全清楚。已有研究[20]证实氨和谷氨酰胺具有神经毒性作用,它们可以导致脑水肿和细胞死亡。这一点可以解释上述UCD病人脑DWI呈高信号表现。
MLD是由于芳香硫酶A或其协同因子活性降低引起的,导致硫酸半乳糖神经酰胺不能降解为半乳糖脑苷脂和硫酸[21-22]。该病常导致中枢神经系统的脑白质(侧脑室周围和半卵圆中心)脱髓鞘,而皮质下白质早期不受累,T2WI上双侧侧脑室周围和半卵圆中心呈对称性高信号。文献[23]报道半卵圆中心在T2WI上高信号区域有“虎纹”或“豹斑”征,而这种线条样或点状结构在增强扫描T1WI上更明显。这种线条或结构在病理上被证实为血管鞘周围残存或未脱髓鞘的正常白质[24]。本研究报道的该例病人侧脑室旁白质、半卵圆中心T2WI上呈高信号,且在侧脑室旁正常和异常的髓鞘呈条纹状交错排列,符合文献报道的“虎纹”征。
总之,神经系统遗传代谢病缺乏特异的临床及影像表现,导致该组疾病容易漏诊、误诊。了解该组疾病的影像表现对缩小鉴别诊断范围有重要作用。早期诊断、早期治疗可以改善患儿预后。
[1]Spronsen FJ,Smit GP,Erwichb JJ,et al.Inherited metabolic diseasesand pregnancy[J].BJOG,2005,112:2-11.
[2]HanL S,Huang Z,Han F,et al.Clinical features and MUT gene mutationspectrumin43Chinesepatientswithisolated methylmalonic acidemia:identification of 10 novel allelic variants [J].World J Pediatr,2015,11:358-365.
[3]刘玥,阴捷,彭芸,等.56例儿童甲基丙二酸血症的脑改变影像学特点[J].医学影像学杂志,2013,23:165-169.
[4]陈娟,李玉华.甲基丙二酸尿症及丙酸尿症的颅脑MRI研究[J].放射学实践,2008,23:595-597.
[5]Radmanesh A,Zaman T,Ghanaati H,et al.Methylmalonicacidemia:brain imaging findings in 52 children and a review of the literature [J].Pediatr Radiol,2008,38:1054-1061.
[6] Baker EH,Sloan JL,HauserNS,et al.MRI characteristics of globuspallidus infarcts in isolated methylmalonic academia[J].AJNR,2015,36:194-201.
[7]Weisfeld-Adams JD,Bender HA,Miley-Åkerstedt A,et al.Neurologic and neurodevelopmental phenotypes in young children with early-treated combined methylmalonic acidemia and homocystinuria,cobalamin C type[J].Mol Genet Metab,2013,110:241-247.
[8]Klee D,Thimm E,Wittsack HJ,et al.Structural white matter changes in adolescents and young adults with maple syrup urine disease[J].J Inherit Metab Dis,2013,36:945-953.
[9] Li X,Ding Y,Liu Y,et al.Eleven novel mutations of the BCKDHA,BCKDHB and DBT genes associated with maple syrup urine disease in the Chinese population:report on eight cases[J].Eur J Med Genet,2015,58:617-623.
[10]Kilicarslan R,Alkan A,Demirkol D,et al.Maple syrup urine disease:diffusion-weighted MRI findings during acute metabolic encephalopathic crisis[J].Jpn J Radiol,2012,30:522-525.
[11]Myers KA,Reeves M,Wei XC,et al.Cerebral edema in maple syrup urine disease despite newborn screening diagnosis and early initiation of treatment[J].JIMD Rep,2012,3:103-106.
[12]Xia W,Yang W.Diffusion-weighted magnetic resonance imaging in a case of severe classic maple syrup urine disease[J].J Pediatr Endocrinol Metab,2015,28:805-808.
[13]Harper PA,Healy PJ,Dennis JA,et a1.Maple syrup urine disease (branchedchain ketoaciduria)[J].Am J Pathol,1990,136:1445-1447.
[14]王宏伟,王晓明,郭启勇,等.新生儿枫糖尿病脑病一例[J].中华放射学杂志,2008,42:1221-1222.
[15]Righini A,Ramenghi LA,Parini R,et al.Water apparentdiffusion coefficient and T2 changes in the acute stage of maple syrupurine disease:evidence of intramyelinic and vasogenic-interstitial edema [J].J Neuroimaging,2003,13:162-165.
[16]叶锦棠,谢晟,齐朝月,等.婴幼儿甲基丙二酸尿症的MR表现[J].中国医学影像技术,2008,24:1192-1194.
[17]Helman G,Pacheco-Colón I,Gropman AL.The urea cycle disorders [J].Semin Neurol,2014,34:341-349.
[18]罗芳,陈正,马晓路,等.鸟氨酸氨甲酰基转移酶缺陷症头颅影像学及脑功能监测:1例报告[J].中国当代儿科杂志,2011,13:165-167.
[19]Takanashi J,Barkovich AJ,Cheng SF,et al.Brain MR imaging in neonatal hyperammonemic encephalopathy resulting from proximal urea cycle disorders[J].AJNR,2003,24:1184-1187.
[20]Bireley WR,Van Hove JL,Gallagher RC,et al.Urea cycle disorders:brain MRI andneurological outcome[J].PediatrRadiol,2012,42:455-462.
[21]Groeschel S,Kehrer C,Engel C,et al.Metachromaticleukodystrophy:natural course of cerebral MRI changes in relation to clinical course [J].J Inherit Metab Dis,2011,34:1095-1102.
[22]程爱兰,金彪.1H-MRS在遗传性脑白质营养不良中的研究进展[J].国际医学放射学杂志,2015,38:9-12.
[23]肖江喜,杨开颜,王霄英,等.儿童异染性脑白质营养不良的MRI表现[J].中华放射学杂志,2011,35:747-750.
[24]Groeschel S,í Dali C,Clas P,et al.Cerebral gray and white matter changes and clinical course in metachromatic leukodystrophy[J]. Neurology,2012,79:1662-1670.
(收稿2015-07-09)
Brain imaging characteristics of inherited metabolic diseases in infants and young children
CHENG Ailan,FENG Yun,HOU Liang,CAO Wenjun,WANG Dengbin,JIN Biao.Department of Radiology,Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine,Shanghai 200092,China
ObjebtiveTo explore the characteristics of brain MRI images in pediatric patients with inherited metabolic diseases.MethodsSixteen pediatric patients within herited metabolic diseases were recruited from January 2014 to December 2014.Their clinical features,laboratory examsand brain MRI were analyzed.ResultsThere were twelve cases of organic academia,eight case of methylmalonicacademia,four cases of maple syrup urine disease.Three cases of urea cycle disorders,and 1 case of metachromatic leukodystrophy.Among the 12 patients with organic acidemia,basal ganglia showed hypointensity on T1FLAIR,mild hyperintensity on T2FLAIR,and intense hyperintensity on DWI(n=7);brainstem and cerebellum showed intense hyperintensity on DWI(n=4 and 2,respectively);corpus callosum showed hyperintensityon DWI in 1 case;brain atrophy in 2 cases;one case showed hypointensity on T1FLAIR,hyperintensity on T2FLAIR,and hyperintensity on DWI atbilateral centrum-semiovale;and ventricular enlargement in 1 case.Among the 3 patients with urea cycle disorders,ventricular enlargement was seen in 2 cases,cerebral cortex showed diffuse hypeninternsity on DWI in 1 case.The case with metachromatic leukodystrophy showed hyperintensity on T2FLAIR at bilateral preventricular white matter and centrum-semiovale.ConclusionThe brain MRI is lack of specificity in infants and young children with inherited metabolic diseases.Different disease types show varied MRI manifestations,and the organic acidemia in children mainly shows basal ganglia involvement.
Inherited metabolic disease;Brain;Magnetic resonance imaging;Methylmalonic academia;Maple syrup urine disease
金彪,E-mail:kking1105@163.com
10.19300/j.2016.L3657
R725.8;R445.2
A
上海交通大学医学院附属新华医院放射科,上海200092
猜你喜欢
老年医学研究(2021年6期)2021-03-09
当代水产(2019年11期)2019-12-23
中国妇幼健康研究(2019年2期)2019-03-26
中成药(2018年5期)2018-06-06
农业环境科学学报(2017年2期)2017-03-20
中外医疗(2015年18期)2016-01-04
转化医学电子杂志(2015年4期)2015-12-27
磁共振成像(2015年1期)2015-12-23
郑州大学学报(医学版)(2015年2期)2015-02-27
中国茶叶加工(2015年3期)2015-02-27
