
甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的合成及结构表征
——推荐一个综合药物合成实验
2016-07-27 09:07:01刘美艳曾佑林湖南师范大学药物合成实验教学中心长沙410081
大学化学 2016年4期
刘美艳 曾佑林(湖南师范大学药物合成实验教学中心,长沙410081)
·化学实验·
甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的合成及结构表征
——推荐一个综合药物合成实验
刘美艳 曾佑林*
(湖南师范大学药物合成实验教学中心,长沙410081)
摘要:介绍一个综合药物合成实验——甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的合成及结构表征。以D-甘露糖为原料,经费歇尔糖苷化和苯甲酰化合成甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷。用薄层色谱监测反应进程,采用过滤、洗涤、浓缩、萃取、干燥、柱层析等操作对中间体和目标产物进行分离纯化,并通过熔点和旋光度测定、核磁共振、红外光谱对其结构进行表征。
关键词:反应进程监测;D-甘露糖结构表征;药物合成实验
药物合成实验是制药工程专业的重要教学内容之一。该课程以强化学生实验操作技能、全面系统地训练和提高学生的综合能力、使学生初步具备独立从事新药开发和研究的能力为培养目标。在药物合成实验开课之前,学生已经完成了无机化学实验、分析化学实验、有机化学实验、物理化学实验、仪器分析实验、化工基础实验等基础实验课程的学习,并已掌握了药物合成实验所需的基本知识和技能。因此,药物合成实验内容不应是基础实验的重复,而应以综合性和专业性为甄选依据,突出基础知识和技能在药物开发和研究领域的综合应用,着力培养学生分析、解决实际问题的能力。为实现培养制药工程专业学生的独立科研与独立工作能力,开设综合性药物合成实验作为药物合成实验教学改革的一项新措施,已越来越受到各高校药物合成实验教学工作者的重视[1-4]。
寡糖是许多药物的重要结构单元,含糖药物已涵盖了抗生素类、心脑血管类、抗肿瘤类、激素类等19个门类[5],临床药物达132种。寡糖结构的多样性与复杂性,增加了含糖药物全合成难度。含糖药物的全合成,取决于寡糖的高立体选择性、高区域选择性、高收率合成,涉及有机合成化学、糖化学、保护基化学、分离过程化学、分析化学等学科知识[6]。笔者结合在寡糖合成领域所取得的经验,经反复筛选、实践,将甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的合成转化为综合性药物合成实验。该综合性实验的设计体现了药物合成研究(合成→分离纯化→结构表征)的特点,不仅可使学生掌握药物合成基本技能以及常见的分离纯化方法和化合物结构表征方法,还能提高学生综合应用化学基本原理和实验技能解决实际问题的能力。
1 实验目的
①了解药物合成过程中的保护基化学。
②巩固回流、过滤、萃取、浓缩等基本操作。
③掌握旋转蒸发仪、旋光仪、红外光谱、核磁共振的原理与使用方法。
④掌握薄层色谱和柱色谱的基本原理,利用薄层色谱法示踪有机反应进程,利用柱层析法分离纯化目标产物。
⑤掌握有机化合物结构表征的常用方法,如红外光谱法和核磁共振法;学会分析红外图谱、1H NMR和13C NMR一维谱、H-H相关和C-H相关二维谱。
⑥利用比旋光度和核磁偶合常数定性表征化合物的立体结构。
2 实验原理
①薄层色谱、柱色谱的基本原理与操作(略)。
②红外光谱、核磁共振、旋光仪的基本原理与操作(略)。
③合成原理:D-甘露糖在无水酸性条件下经费歇尔糖苷化反应,立体选择性合成甲基α-D-吡喃甘露糖苷。反应混合物经过滤、洗涤等后处理过程分离纯化得到精品甲基α-D-吡喃甘露糖苷。以提纯后的甲基α-D-吡喃甘露糖苷为原料,以吡啶为反应溶剂和缚酸剂,以苯甲酰氯为酰化试剂进行酯化反应,对甲基α-D-吡喃甘露糖苷的四个羟基进行保护。通过水解(或醇解)破除过量的苯甲酰氯,通过浓缩除去大部分吡啶,通过稀盐酸(或饱和硫酸铜溶液)洗涤除去少部分吡啶和大部分吡啶盐酸盐,通过柱层析得到精品甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷。合成路线如图1所示。
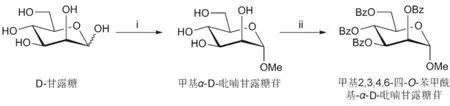
图1 甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的合成路线
3 试剂和材料
D-甘露糖,无水甲醇,乙酰氯,苯甲酰氯,吡啶,二氯甲烷,饱和硫酸铜溶液(或10%盐酸溶液),无水硫酸钠,4-N,N-二甲氨基吡啶(DMAP),二次蒸馏水,氯仿,氘代氯仿,重水,显色剂:30%硫酸-乙醇溶液,展开剂(A):V乙酸乙酯:V甲醇=5:3,展开剂(B):V乙酸乙酯:V石油醚=1:4,洗脱剂:V乙酸乙酯:V石油醚=1:5,HF254薄层色谱板(规格2.5 cm×5.0 cm,100片左右,保存在干燥器中),层析硅胶(200-300目)。
4 仪器设备
CL-2型集热式磁力搅拌器,回流冷凝管,搅拌磁子,铁架台,烧瓶夹,冷凝管夹,抽滤瓶,布氏漏斗,100 mL三颈烧瓶,100 mL单口茄形烧瓶,100 mL玻璃层析缸,19#空芯塞,显色剂雾化器,培养皿(内径15 cm),玻璃刀,100 mL量筒,10 mL量筒,2 mL刻度滴管,滤纸,剪刀,铅笔,直尺(学生自带),封闭式电炉,循环水式多用真空泵,三用紫外灯(2台),旋转蒸发器,层析柱(d=3.0 cm,l=40 cm),500 mL溶剂球,玻璃棒(80 cm),脱脂棉,铝质试管架,试管(d=1.2 cm,l=15 cm),漏斗,烧杯,药用熔点仪,封端熔点管,旋光仪,红外光谱仪,核磁共振仪,核磁样品管,50 mL容量瓶。
5 实验进度安排
实验分4个阶段进行,具体安排如下:
第一阶段为甲基α-D-吡喃甘露糖苷的合成。教师利用反应等待时间讲解薄层色谱与柱色谱的基本原理、薄层色谱法示踪反应进程的操作方法。
第二阶段为甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的合成。教师利用反应等待时间讲解有机化合物结构表征常见方法的基础知识。
第三阶段为柱色谱分离纯化甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷。
第四阶段为甲基-α-D-吡喃甘露糖苷和甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的比旋光度及红外光谱的测定以及甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的1H NMR、13C NMR一维谱、H-H相关和C-H相关二维谱的测定。教师对结构表征结果进行解析。该阶段实验按如下方案实施:①组织学生参观核磁共振实验室和红外光谱室,简要介绍实验室情况;②将学生分成A、B两组,A组先在一名教师指导下完成甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的核磁共振实验,再在另一教师指导下完成熔点测定、比旋光度测定和红外光谱的实验;B组先在一名教师指导下完成甲基α-D-吡喃甘露糖苷的熔点测定、比旋光度测定和红外光谱的实验,再在另一教师指导下完成核磁共振实验;③汇总实验结果,对结构表征结果进行解析。
6 实验步骤
6.1 第一阶段
在干燥的100 mL三颈烧瓶中加入搅拌磁子和无水甲醇(10 mL),然后将其安装至集热式磁力搅拌器上。室温搅拌下缓慢滴加乙酰氯(1 mL,用刻度滴管滴加)。然后一次性加入D-甘露糖(3.0 g,16.7 mmol),装上回流冷凝管,侧口用空芯塞塞上,加热回流反应。用TLC(展开剂:V乙酸乙酯:V甲醇= 5:3,显色方法为硫酸碳化法)监测反应(每小时监测一次)至原料基本消失,3小时左右反应基本完全,此时反应混合物为固液混合物。冷却至室温,过滤,抽干得白色固体(若产品带颜色,可用少量甲醇洗涤至白色)。用TLC检验产品纯度合格(单一斑点)固体称重后,将各组产品合并。将各组滤液合并,浓缩至1/2体积,有固体析出,过滤得白色固体,该固体经TLC纯度检测合格后,将两部分产品合并,真空干燥后备用。
6.2 第二阶段
称取甲基α-D-吡喃甘露糖苷(0.5 g,2.57 mmol)置于100 mL单口茄形瓶中,加入吡啶(10 mL)和催化剂DMAP(50 mg,也可不加)。室温搅拌下缓慢加入苯甲酰氯(1.5 mL,12.6 mmol),加完让其继续反应。用TLC(V石油醚:V乙酸乙酯=3:1,先在紫外灯照射下用铅笔描出荧光斑点,再用硫酸碳化显色)监测。若1.5小时仍未反应完全,可通过加热(70°C)促进反应;若加热半小时仍未反应完全,可补加少量苯甲酰氯。待TLC监测反应完全后,加入少量甲醇(约1 mL,若无甲醇,可加1 mL水)反应数分钟。浓缩去除溶剂,往烧瓶中依次加入自来水(50 mL)和二氯甲烷(50 mL)将残留物溶解,将混合物转入分液漏斗中,分液。有机层依次用10%稀盐酸水溶液(约100 mL,也可用饱和硫酸铜溶液)、自来水(约150 mL)萃取。有机层用无水Na2SO4干燥。过滤,将各组滤液合并上交教师保存备用。
6.3 第三阶段
将玻璃层析柱固定在铁架台上,下端用烧瓶夹托住,上端用冷凝管夹固定。在层析柱底部填充少量脱脂棉,并用玻璃棒压住,再往层析柱中加适量洗涤剂(高度约5 cm),铁架台上放一250 mL烧杯,用于收集流出液。另取适量硅胶置于烧杯(500 mL,可几组共用)中,加洗脱剂将硅胶浸湿。然后将硅胶转至层析柱中,使硅胶高度达到约20 cm。将层析柱中液面放低至与硅胶层相切,然后用双手轻轻拍击层析柱中部,此时硅胶层下降(若硅胶层高度不够20 cm,应补加硅胶)。将第二阶段实验所得有机溶液浓缩(使残留浆状物具有一定流动性,太黏或太稀均影响柱分离效果),然后将层析柱中液面放低至与硅胶层相切,用滴管将残留浆状物转移至层析柱中,使残留浆状物厚度为5 mm(注意:尽量将残留浆状物送至硅胶上水平面,不要将硅胶冲起),并覆盖整个硅胶。打开阀门,将残留浆状物上水平面放低至与硅胶层相切,关闭阀门,在层析柱中加入2-3 cm厚的无水硫酸钠(食盐亦可)。然后向层析柱中加洗脱剂使液面接近层析柱上口部位,打开阀门,套上溶剂球,再往溶剂球中加更多的洗脱剂。此时用试管收集流出液,并用TLC监测流出液组成成分情况。(注意:最初不需将TLC板展开,只要观察是否有荧光斑点出现;当出现荧光斑点时,此时将TLC板展开。)当TLC显示为单一目标产物时(由教师确定),合并该部分洗脱液(可与其他组合并),浓缩回收产品及洗脱剂,其他洗脱液直接回收。当流出液不含单一目标产物时,停止柱层析操作,并回收层析柱内的洗脱剂。所有产物浓缩后,真空干燥备用。
6.4 第四阶段
①甲基α-D-吡喃甘露糖苷的比旋光度测定。
准确称取甲基α-D-吡喃甘露糖苷(0.5000 g),用二次蒸馏水溶解并定容至50 mL。用二次蒸馏水做背景消除,再测定样品的旋光度,复测4次,求5次测定的平均值。平均值乘以100即为甲基α-D-吡喃甘露糖苷的比旋光度。
②甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的比旋光度测定。
以氯仿为溶剂,按照甲基α-D-吡喃甘露糖苷的比旋光度测定方法进行测定。
③甲基α-D-吡喃甘露糖苷的红外光谱测定。
将溴化钾研细后放置于红外灯下,取少量甲基α-D-吡喃甘露糖苷与溴化钾研磨混匀,并在红外灯下烘烤数分钟。压制背景片和样品片,用背景片做背景消除后,测定样品红外光谱。
④甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的红外光谱测定。
按照甲基α-D-吡喃甘露糖苷的红外光谱测定方法进行测定。
⑤甲基α-D-吡喃甘露糖苷的核磁共振谱测定。
往核磁管中加入适量甲基α-D-吡喃甘露糖苷(约10-20 mg)和重水(0.5 mL),用核磁共振仪测定甲基α-D-吡喃甘露糖苷的1H NMR、13C NMR一维谱、H-H相关和C-H相关二维谱。
⑥甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的核磁共振谱测定。
以氘代氯仿(0.5 mL)为溶剂,测定甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的1H NMR、13C NMR一维谱、H-H相关和C-H相关二维谱。
⑦甲基α-D-吡喃甘露糖苷的熔点测定。
⑧甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的熔点测定。
7 结果与讨论
7.1 红外光谱
7.1.1 甲基α-D-吡喃甘露糖苷的红外光谱
由图2可知,3455 cm-1是O―H伸缩振动峰(分子间氢键),3293 cm-1是O―H伸缩振动峰(缔合),2946 cm-1是C―H伸缩振动峰,1403 cm-1是C―H弯曲振动峰,1122、1033、971 cm-1是C―O伸缩振动峰。
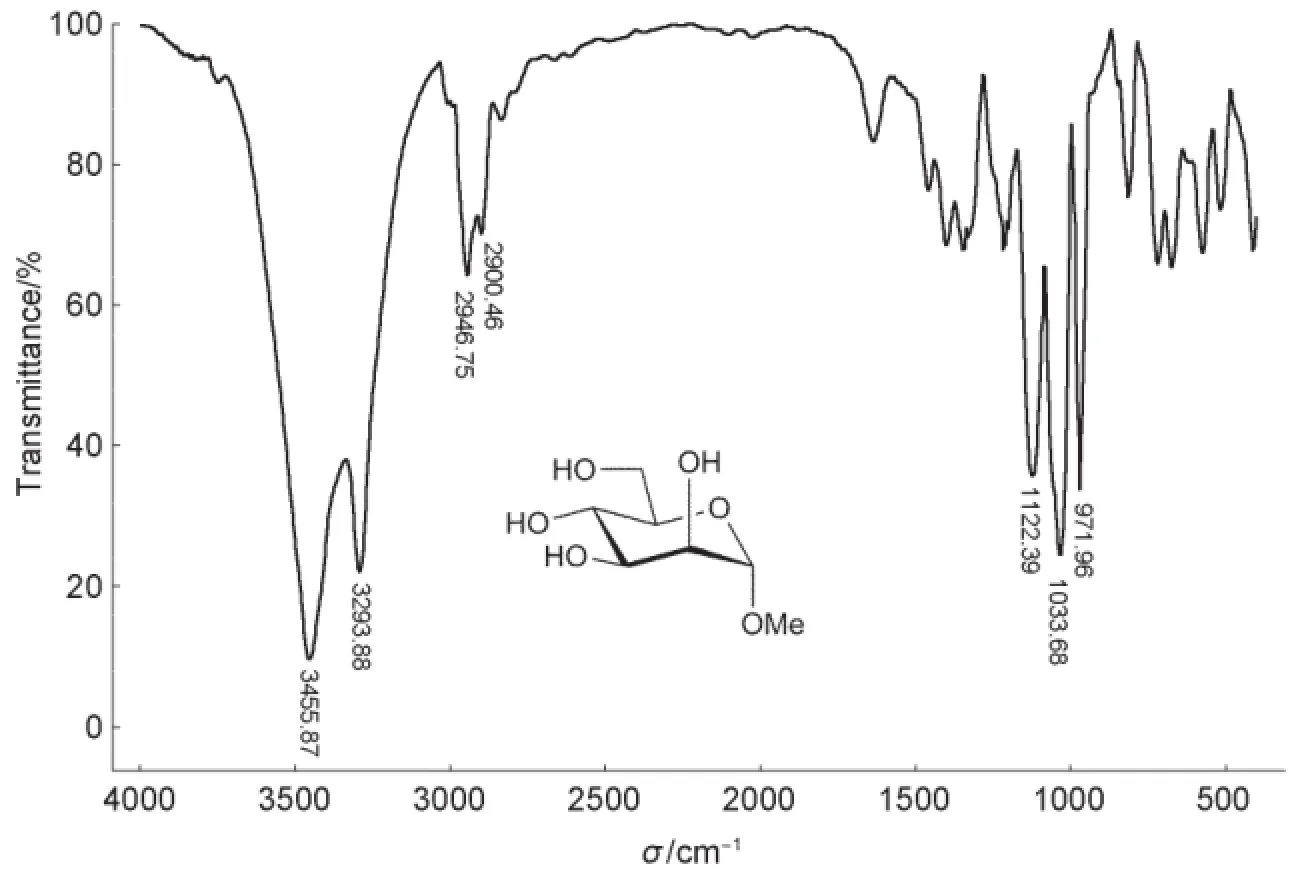
图2 甲基α-D-吡喃甘露糖苷的红外光谱图
7.1.2 甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的红外光谱
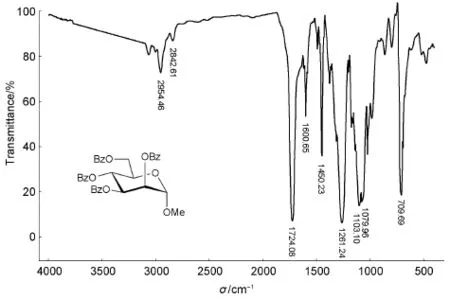
图3 甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的红外光谱图
由图3可知,2954、2842 cm-1是C―H伸缩振动峰,1724 cm-1是C=O伸缩振动峰,1600-1450 cm-1是苯环骨架振动峰,1261、1103、1079 cm-1是C―O伸缩振动峰,709 cm-1是单取代苯环上的C―H面外弯曲振动峰。
对比图2和图3,C=O伸缩振动峰和苯环骨架振动峰证实羟基已被苯甲酰化。
7.2 核磁共振、旋光度和熔点
甲基α-D-吡喃甘露糖苷和甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的核磁共振、旋光度和熔点数据如下:
由1H NMR可知,3.30处为3个氢的单峰,为甲基氢,说明甘露糖成功甲基化。耦合常数J1,2=1.5 Hz和1JC-1,H-1=172.0 Hz共同说明糖苷键为a键,即糖苷化产物为α-异构体。
②甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷[α=-66.2°(c 1,HCCl3)。1H NMR(500 MHz,DCCl3):δ 8.15-7.82(m,8H,4 o-Ph-H),7.61-7.25(m,12H,4 m-and p-Ph-H),6.12(dd,1H,J4,3=J4,5=10.2 Hz,H-4),5.92(dd,1H,J3,4=10.2 Hz,J3,2=3.0 Hz,H-3),5.71(dd,1H,J1,2=1.0 Hz,J2,3=3.0 Hz,H-2),5.01(d,1H,J1,2=1.0 Hz,H-1),4.72(dd,1H,J6a,5=2.5 Hz,J6a,6b=12.0 Hz,H-6a),4.52(dd,1H,J6b,5=4.5 Hz,J6b,6a=12.0 Hz,H-6b),4.42(ddd,1H,J4,5=10.2 Hz,J5,6a=2.5 Hz,J5,6b=4.5 Hz,H-5),3.55(s,3H,H-OMe)。13C NMR(125 MHz,DCCl3):δ 166.2,165.5,165.4 (4C,4 PhCO-C,one signal overlapped),133.5,133.2,133.1,129.9,129.8,129.7,129.3,129.1,129.0,128.6,128.5,128.3(24 C,4 Ph-C,some signals overlapped),98.7(1C,1JC-1,H-1=174.1 Hz,C-1),70.4(1C,C-2),70.1(1C,C-3),68.8(1C,C-5),66.9(1C,C-4),62.9(1C,C-6),55.6(1C,C-OMe)。m.p.133.9-134.8°C。
1H NMR中8.15-7.25区间内有20个氢,为4个苯甲酰基苯环上的氢;13C NMR中166.2、165.5、165.4处共有4个C,为苯甲酰基的羰基碳,说明甲基α-D-吡喃甘露糖苷的四个羟基均成功酯化。偶合常数J1,2=1.0 Hz和1JC-1,H-1=174.1 Hz共同说明糖苷键为a键,即目标产物为α-异构体。
8 思考题
(1)将醛或酮转化为相应的缩醛或缩酮的反应条件应为干燥氯化氢气体的醇溶液,费歇尔糖苷化的实质是半缩醛羟基醚化形成缩醛,为何采用甲醇与乙酰氯就能实现甘露糖的糖苷化?
(2)甘露糖的甲基苷化的主要产物是α-异构体,β-异构体为痕量产物。从反应机理和空间电子效应角度出发,对甘露糖的甲基苷化反应的立体选择性进行解释。
(3)如何用薄层色谱监测反应进程,判断费歇尔糖苷化和苯甲酰化反应是否完全(或达到平衡状态)的依据分别是什么?如何促使苯甲酰化反应更彻底?
(4)如何选择柱层析的流动相?
(5)影响柱层析分离效果的因素有哪些?
(6)在测定物质的比旋光度时,背景消除时仪器示数非常稳定,但测定样品时出现仪器示数不稳定现象,如何解决?
(7)以氘代氯仿为核磁溶剂时,为何13C NMR谱图在77.3处有三重峰(溶剂峰)?若以氘代二甲亚砜为核磁溶剂,则13C NMR谱图中溶剂峰是几重峰?
(8)甲基α-D-吡喃甘露糖苷的分子式为C7H14O6,但从1H NMR谱图获知氢的积分值之和为10,缺失4个氢,试解释其原因?
(9)如何确定中间体和目标产物均为α-异构体?
(10)如何对核磁共振谱图中的氢或碳进行归属,如何计算偶合常数?
(11)dd峰、t峰(三重峰)、q峰(四重峰)之间有何区别与联系?
9 结论
本文介绍的甲基2,3,4,6-四-O-苯甲酰基-α-D-吡喃甘露糖苷的合成及结构表征实验源于科学研究,涵盖了药物合成研究的合成、分离纯化、结构表征3个重要环节。该实验内容新颖,所用药品、试剂和材料便宜易得,实验重现性好,不仅适合制药工程专业实验教学,也适合于有机合成专业实验教学。通过实验,能加深学生对多步合成、官能团保护、立体选择性和区域选择性、反应进程监测与判断、薄层色谱和柱色谱的应用、结构表征等相关知识的理解,也能提高学生的实验技能、科研兴趣和创新能力。本实验已在湖南师范大学化学化工学院制药工程专业本科生的药物合成实验课程中进行了5年的教学实践,取得了良好的教学效果。
参考文献
[1]徐凌云,赵静国,陈 平,郑卫平,胡明秀.药学教育,2014,30,69.
[2]吴胜昔,胡 勇,陈忠敏.科学咨询(科技·管理),2014,14,127.
[3]刘小平,徐海星,李湘南,申永存,马定桂.实验室科学,2009,69,15.
[4]李元祥,林红卫.广东化工,2010,37,177.
[5]杜晓光,耿美玉.生命科学,2011,23,671.
[6]孔繁祚.糖化学.北京:科学出版社,2005.
中图分类号:O6-3;G64
doi:10.3866/PKU.DXHX20160460www.dxhx.pku.edu.cn
*通讯作者,Email:youlinzengcn@gmail.com
基金资助:国家自然科学基金面上项目(21272064);湖南省教育厅资助科研项目(12C0249);湖南师范大学实习基地项目(043-008)
Synthesis and Structural Characterization of Methyl 2,3,4,6-tetra-O-benzoyl-α-D-mannopyrannoside
LIU Mei-Yan ZENG You-Lin*
(Drug Synthesis Experiment Teaching Center,Hunan Normal University,Changsha 410081,P.R.China)
Abstract:A new comprehensive drug synthesis experiment was introduced in this article.From commercially available D-mannose,methyl 2,3,4,6-tetra-O-benzoyl-α-D-mannopyrannoside was synthesized via Fischer-Helferich glycosylation followed by benzoylation.The intermediate and the target compound were purified by filtration,washing,concentration,extraction,drying,and silicon column separation.The structure was characterized by the infrared spectrum(IR),nuclear magnetic resonance (NMR)spectrum,measurements of melt point and optical rotation of the compound.
KeyWords:Reactionmonitoring;D-mannose;Structuralcharacterization;Drugsynthesisexperiment
