
两个组氨酸希夫碱镍(Ⅱ)配合物的合成、晶体结构和DNA相互作用及SOD活性
2016-07-22 08:26:48董建方李文彬赵培然丁菲菲李连之聊城大学化学化工学院聊城5059山东工程技师学院材料科学系聊城507
无机化学学报 2016年5期
魏 强 董建方李文彬 赵培然 丁菲菲 李连之(聊城大学化学化工学院,聊城 5059)(山东工程技师学院材料科学系,聊城 507)
两个组氨酸希夫碱镍(Ⅱ)配合物的合成、晶体结构和DNA相互作用及SOD活性
魏强1董建方2李文彬1赵培然1丁菲菲1李连之*,1
(1聊城大学化学化工学院,聊城252059)
(2山东工程技师学院材料科学系,聊城252027)
摘要:合成了2个新的L-组氨酸水杨醛希夫碱混配体镍(Ⅱ)配合物,[Ni(Sal-L-His)(Phen)]·H2O(1)(Sal-L-His=L-组氨酸水杨醛希夫碱,Phen=菲咯啉)和[Ni(Sal-L-His)(Bipy)]2·2CH3OH·3H2O(2)(Bipy=2,2′-联吡啶)。X射线单晶衍射测定表明,1为单核配合物,属于单斜晶系,P21/c空间群,晶胞参数为:a=1.586 31(13)nm,b=1.215 11(11)nm,c=1.211 30(9)nm,β=106.581(2)°,V=2.237 7(3)nm3Z=4。2为双核配合物,属于正交晶系,C2221空间群,晶胞参数为:a=1.415 30(13)nm,b=1.717 71(15)nm,c=2.256 02(19)nm,V 5.484 6(8)nm3,Z=4。紫外吸收光谱、荧光光谱、圆二色光谱和粘度测定等研究表明,2个配合物与小牛胸腺DNA(CT-DNA)均以插入方式相结合。运用NBT光照还原法测定了配合物的超氧化物歧化酶(SOD)活性,求得IC50(1)=51.5 μmol·L-1,IC50(2)=58. μmol·L-1。
关键词:镍配合物;L-组氨酸;希夫碱;晶体结构;DNA;SOD
山东省自然科学基金(No.Y2004B02)和聊城大学大学生科技文化创新基金(No.SF2014042)资助项目。
*通信联系人。E-mail:lilianzhi1963@163.com;会员登记号:S06N1205M1202。
0引言
希夫碱过渡金属配合物具有抗肿瘤、抗菌、抗癌等多种生物活性[1]。近年来,氨基酸希夫碱过渡金属配合物由于其多方面的性质特别是在医药学中的潜在应用引起人们的极大兴趣[2]。镍作为一种生命必需元素,存在于生物体的系列酶中,比如脲酶、镍铁氢化酶、超氧化物歧化酶等[3-4]。它具有促进红细胞再生,刺激生血功能的作用,在维持大分子结构稳定性、膜稳定性和细胞的超微结构方面也起到重要作用。鉴于镍所特有的生物活性,近来镍及其化合物越来越受到广大科研工作者的关注。文献报道许多镍的配合物表现出良好的抗菌、抗癌、抗氧化以及DNA结合和DNA切割活性[5-6]。
核酸是生物体的重要组成成份,它对生物的生长、发育、繁殖、变异等现象起着决定作用,一直是现代生物学、化学和医学的重要研究领域。DNA作为生命遗传信息的携带者和传递者,是许多药物的主要作用靶点。在分子水平上研究药物或者配合物小分子与DNA等生物大分子的相互作用,是当前生命科学、药物化学的重要课题之一。这对于研究致癌化合物的致癌机理和抗癌药物的药理和毒性以及设计合成新型药物方面都有很大意义[7-8]。超氧阴离子自由基(O2-·)是生物体内有氧代谢的产物,也是对生命体系毒性很高的物种[9],导致包括脂类物质的超氧化、DNA损伤、老化、癌症和许多其他疾病。超氧化物歧化酶(SOD)能有效清除O2-·,在抗辐射损伤、减缓衰老以及预防癌症等方面有其独特作用[10]。天然SOD生产成本高,且寿命短、易失活,分子量大不易通过细胞膜,在应用上受到一定限制。因此,人工合成稳定、无毒、分子量小的活性化合物来替代天然SOD具有重要意义[11]。鉴于此,我们合成了2种新的氨基酸希夫碱镍(Ⅱ)配合物,测定了其晶体结构,利用紫外可见吸收光谱、荧光光谱、圆二色光谱和粘度测量等方法,研究了这两个配合物与小牛胸腺DNA (CT-DNA)的作用,还利用氮蓝四唑(NBT)光照还原法测定了配合物的模拟SOD的性质。
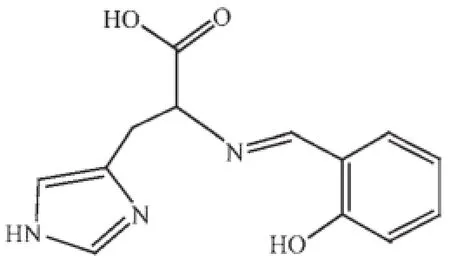
Scheme 1 Structure of the Schiff base ligand
1 实验部分
1.1试剂与仪器
水杨醛购自阿法埃莎化学技术有限公司,L-组氨酸购自北京经科宏达生物技术有限公司,氮蓝四唑(NBT)购自AMRESCO,核黄素购自上海生工生物工程股份有限公司,N,N-四甲基乙二胺购自SIGMA ALDRICH,小牛胸腺DNA(CT-DNA),溴化乙锭(EB)和三羟甲基氨基甲烷(Tris)均购自华美生物工程有限公司。CT-DNA储存溶液用10 mmol·L-1Tris-HCl/ 10 mmol·L-1NaCl,pH 7.1缓冲溶液配制,经UV光谱测定A260/A280>1.8,CT-DNA的浓度通过测量其在260 nm处的吸光度,然后根据其摩尔吸光系数ε260=6 600 L·mol-1·cm-1来确定[12]。其它试剂如四水合醋酸镍、磷酸氢二钠、磷酸二氢钠、菲咯啉、2,2′-联吡啶等均为分析纯试剂。
Bruker Smart-1000 CCD型衍射仪,Nicolet 5700 FT-IR型红外光谱仪 (KBr压片),UV-2500 PC型紫外-可见光光度计(Shimadzu,日本),LS55型荧光光谱仪(Perkin Elmer,美国),JASCO J-810型圆二色光谱仪(Japan),乌氏粘度计。
1.2配合物1和2的合成
取0.155 2 g(1.0 mmol)L-组氨酸和0.056 1 g (1.0 mmol)KOH于圆底烧瓶中,加入10 mL甲醇,加热搅拌至完全溶解后,滴加110 μL(1.0 mmol)水杨醛的甲醇溶液(4 mL)。60℃下搅拌回流1 h后,再滴加0.249 8 g(1.0 mmol)四水合醋酸镍的甲醇溶液(4 mL),继续回流反应2 h。最后,再逐滴加入含0.198 0 g(1.0 mmol)菲咯啉(1)或0.156 2 g(1.0 mmol)2,2′-联吡啶(2)的甲醇溶液(4 mL)。加热回流反应3 h,室温冷却过滤,滤液在室温下静置数天,得到块状单晶。配合物1为棕色块状单晶,产率约为60%。元素分析按 C25H21N5NiO4计算的理论值 (%):C 58.40;H4.12;N13.62;实验值(%):C58.36;H4.08%;N 13.58。配合物1的IR(cm-1):3 421(s,νO-H);1 622(s,νC=N);1 592(s,νCOO);546(w,νNi=O);494(w,νNi-N)。配合物2为浅棕色块状单晶,产率约为58%。元素分析按C48H52N10Ni2O11计算的理论值 (%):C 54.27;H 4.93;N,13.18。实验值(%):C 54.31;H 4.91;N 13.15。配合物2的IR(cm-1):3 424(s,νO-H);1 624(s,νC=N);1 593(s,νCOO);545(s,νNi=O);496(m,νNi-N)。
1.3晶体结构测定
选取尺寸为0.34 mm×0.30 mm×0.22 mm的配合物1和0.13 mm×0.11 mm×0.05 mm的配合物2的单晶,置于Bruker Smart-1000 CCD型单晶衍射仪上,以石墨单色化的Mo Kα(λ=0.071 073 nm)辐射为光源,以φ-ω扫描方式收集衍射点。选取I>2σ(I)的可观察点(1为2 150个,2为2 226个)用于结构解析和修正。衍射数据用SAINT程序进行还原[13],并用SADABS程序对衍射强度数据进行吸收校正[14]。单晶结构由直接法以SHELXS程序获得。结构解析和精修均采用SHELXTL-97软件完成[15]。非氢原子的坐标在差值Fourier合成中确定,并对其坐标及各向异性热参数进行了全矩阵最小二乘法修正。氢原子采用理论加氢得到。配合物的主要晶体学数据列于表1。
CCDC:1038774,1;1420088,2。
1.4配合物与DNA的相互作用
1.4.1紫外可见吸收光谱测定
用10 mmol·L-1Tris-HCl/10 mmol·L-1NaCl,pH 7.1缓冲液配制一系列试样。固定配合物的浓度为15 μmol·L-1,逐渐增大CT-DNA的浓度,反应体系于室温放置1 h。配制等浓度的缓冲溶液或DNA缓冲溶液作为参比,设定波长范围200~500 nm,测定不同DNA浓度下配合物的紫外可见吸收光谱。
1.4.2配合物对DNA-EB体系荧光光谱的影响
用10 mmol·L-1Tris-HCl/10 mmol·L-1NaCl,pH 7.1缓冲液配制一系列试样。固定CT-DNA浓度为15 μmol·L-1,EB浓度为3 μmol·L-1,配合物的浓度逐渐增加,反应体系于室温放置1 h后测定荧光光谱狭缝宽度:10 nm,激发波长为525 nm;发射波长范围为540~680nm,扫描速度:300nm·min-1。
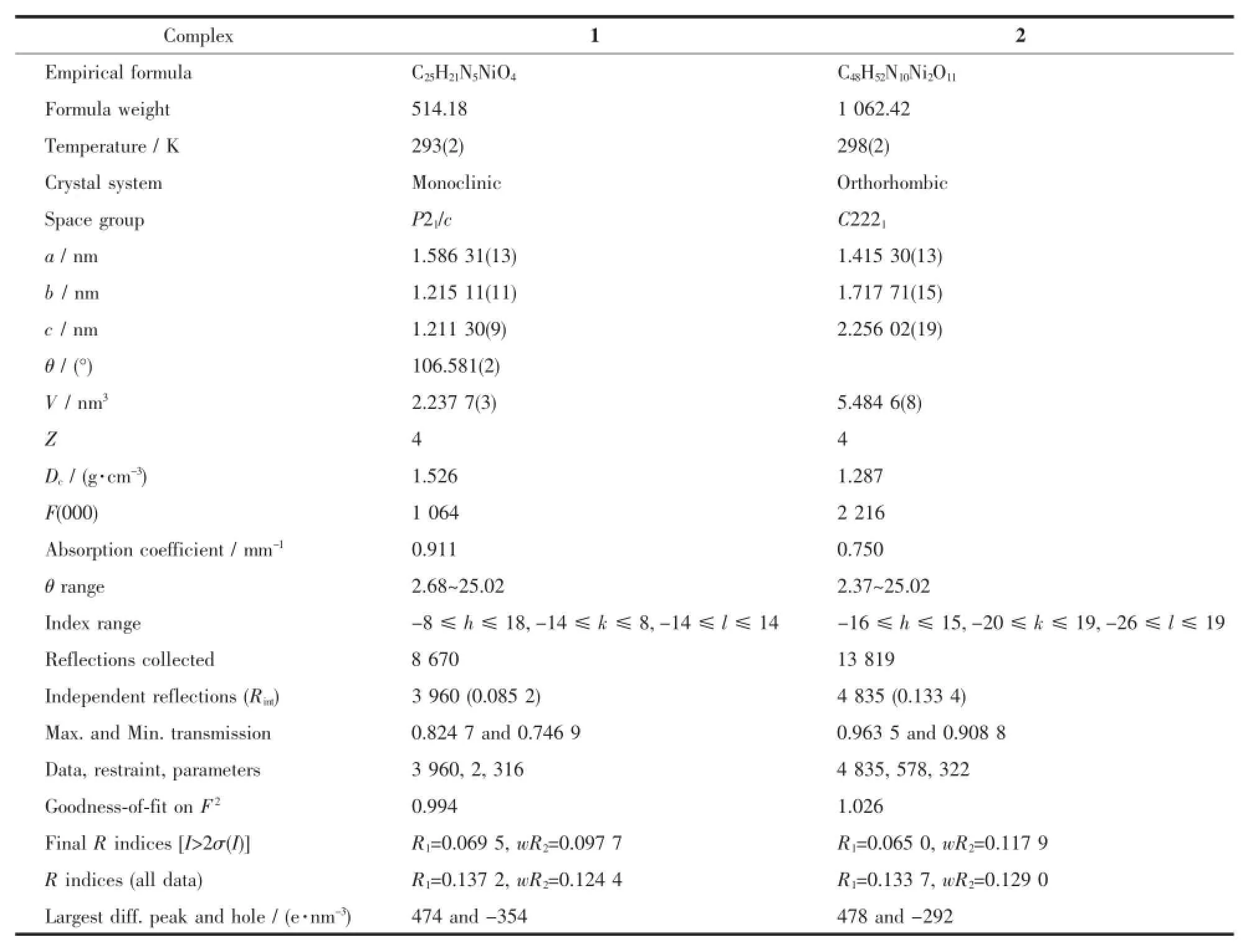
表1 配合物的晶体学数据Table 1 Crystallographic data for the two complexes
1.4.3圆二色光谱测定
用10 mmol·L-1Tris-HCl/10 mmol·L-1NaCl,pH 7.1缓冲液配制一系列试样。固定CT-DNA的浓度为100 μmol·L-1,配合物的浓度逐渐增加,反应体系于室温放置1 h,测定DNA的圆二色光谱(扣除缓冲液和配合物的光谱)。扫描范围为220~320 nm,扫描速度为200 nm·min-1,路径长度1 cm,响应时间为1 s。1.4.4粘度测定
用10 mmol·L-1Tris-HCl/10 mmol·L-1NaCl,pH 7.1缓冲液配制一系列试样。固定CT-DNA的浓度为100 μmol·L-1,逐渐增加配合物浓度,设r=ccomplex/ cDNA,使r分别为0、0.02、0.04、0.06、0.08。试样在(30± 0.1)℃下水浴中恒温30 min后,测定溶液流经毛细管的时间t。每个样品重复测定3次,取平均值。相对粘度η=(t-t0)/t0,以(η/η0)1/3对r作图,t0为缓冲溶液流经毛细管所需的时间,η0为r=0时DNA溶液的相对粘度。在相同条件下,测定EB和甲基绿(MG)对DNA溶液相对粘度的影响。
1.5SOD活性测试
采用NBT光照还原法测定配合物的SOD活性[16]。用pH=7.8的10.0 mmol·L-1磷酸盐缓冲液配制1.0×10-4mol·L-1NBT,6.2×10-6mol·L-1核黄素和8.3×10-4mol·L-1N,N-四甲基乙二胺和配合物(浓度为1.0×10-6~9.0×10-6mol·L-1)溶液。室温下,用恒定光强的冷光灯照射,每光照1 min用紫外可见吸收光谱仪测定其在560 nm处的吸光度,测定时间为10 min。
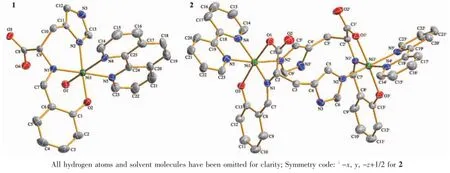
图1 配合物1和2的分子结构(椭球率30%)Fig.1 Molecular structures of complex 1 and complex 2 with thermal ellipsoids at 30%probability
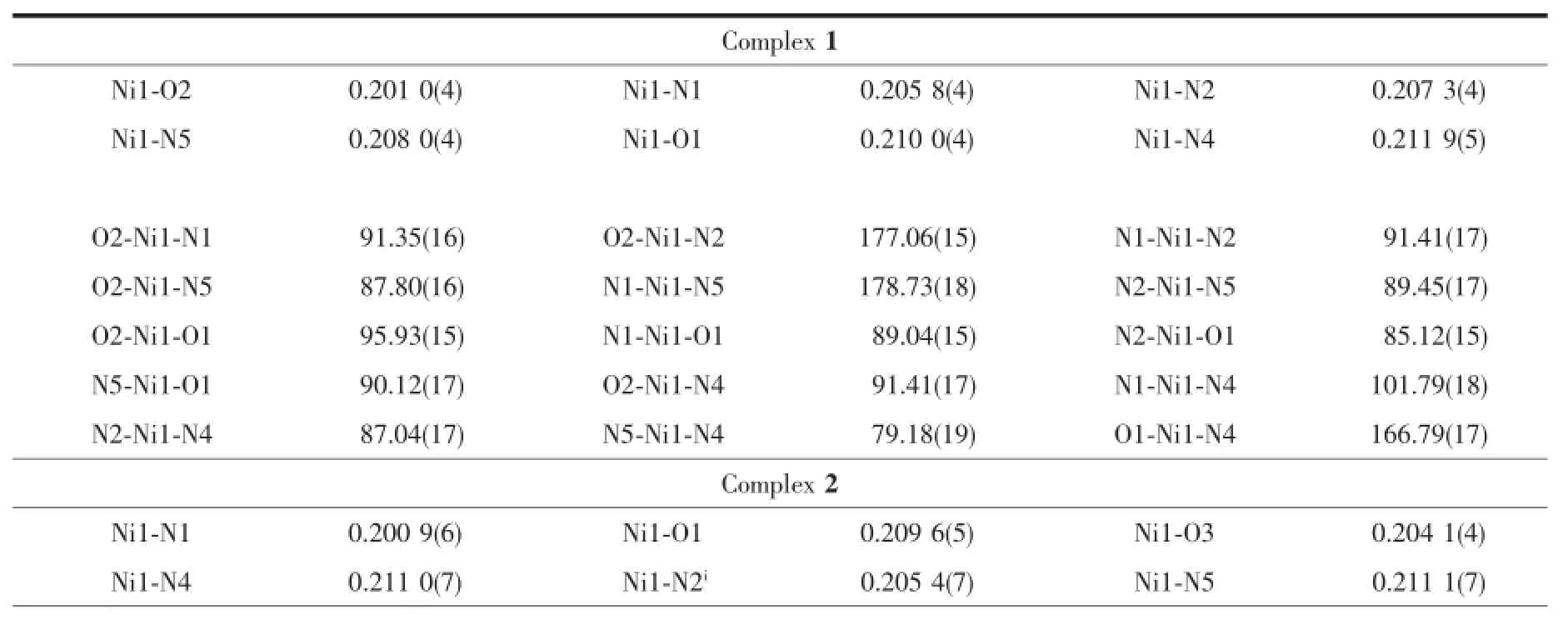
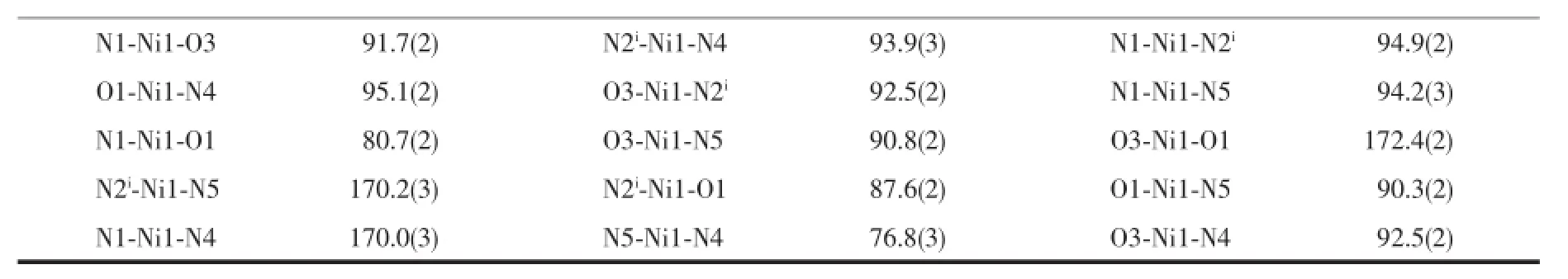
表2 配合物的主要键长和键角Table 2 Selected bond lengths(nm)and angles(°)for the complexes
2 结果与讨论
2.1晶体结构解析
配合物的分子结构见图1,主要键长和键角见表2。在配合物1的分子结构中,Ni(Ⅱ)离子与组氨酸水杨醛希夫碱配体中的酚羟基氧原子O2,亚氨基氮原子N1,咪唑环上的氮原子N2,菲咯啉配体中的2个氮原子(N4和N5)和水分子的氧原子O1配位,形成一个六配位的变形八面体结构。N1,N2,N5和O2原子位于八面体的赤道平面上,O1和N4原子位于八面体的轴向位置,Ni(Ⅱ)离子几乎与赤道平面共面,其偏离基本平面位置距离只有0.000 07(2)nm,O1-Ni1-N4形成的键角为166.79(17)°,表明配位原子围绕Ni(Ⅱ)形成一畸变的八面体。另外菲咯啉平面分子与Ni(Ⅱ)离子配位后,形成一个五元环,其平面与赤道平面之间形成的二面角为84.69(12)°,几乎垂直于赤道平面。值得注意的是,在由氨基酸希夫碱配体参与形成的配合物中,羧基氧原子往往参与金属离子的配位[17-18],而在该配合物中羧基氧原子未参与配位,而是组氨酸咪唑环上的氮原子配位。这可能是由于咪唑环上的氮原子配位更加利于配合物的稳定,这种配位形成一个六元环,若是羧基氧原子配位则形成五元环,其张力要比六元环大些。在配合物1的晶体中,分子间氢键O1-H1D…O3ii和N3-H1…O3i,以及分子内氢键O1-H1C…O4将配合物分子连接形成一个二维平面结构(图2)。其键长和键角见表3。
续表2
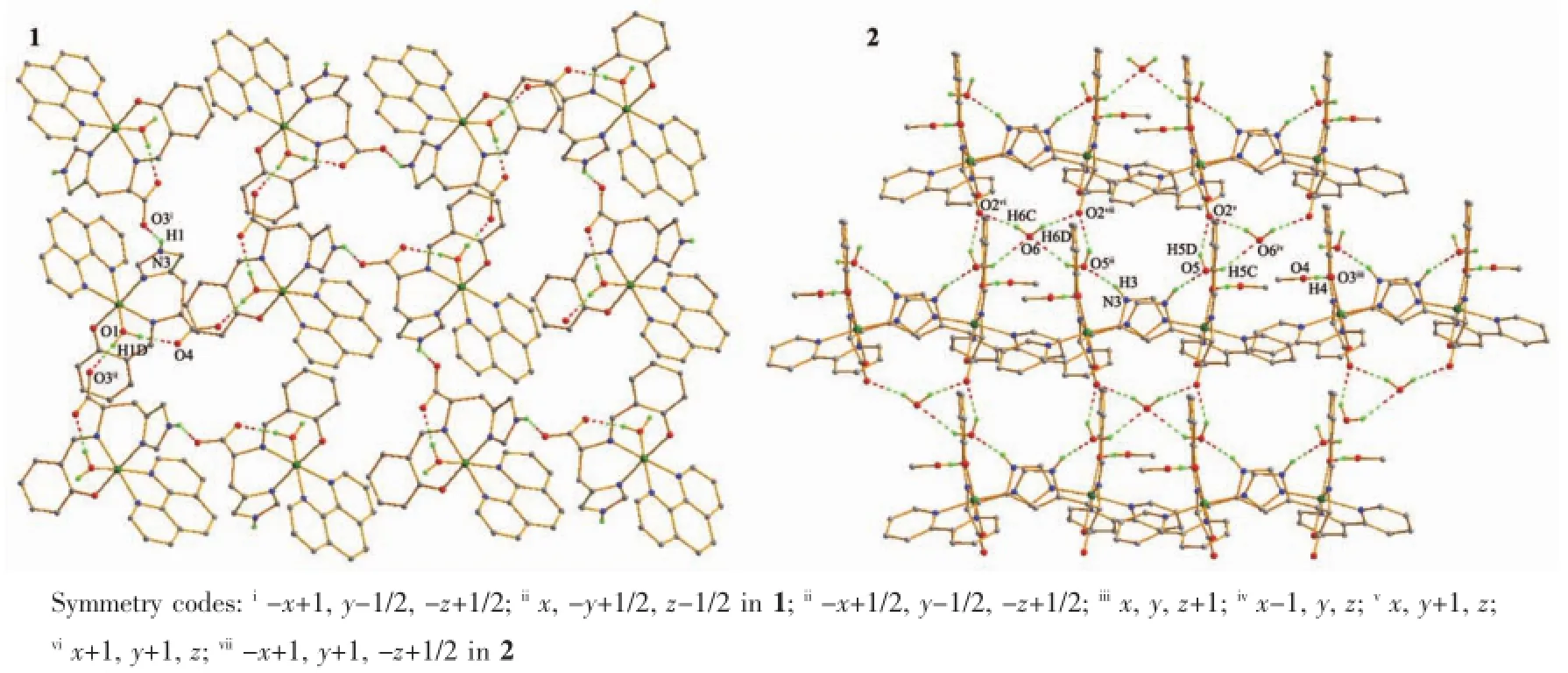
图2 配合物1和2的二维结构Fig.2 2D structure of complex 1 and 2
在配合物2的不对称结构单元中,包含1个希夫碱和菲咯啉混配体双核Ni(Ⅱ)配合物主体分子,2个甲醇和3个水溶剂分子。配合物2主体分子为希夫碱配体上咪唑环桥连的具有中心对称结构的双核配合物。由于分子结构的对称性,这里仅讨论对称单元中的一部分。配合物的分子结构中,Ni(Ⅱ)离子与组氨酸水杨醛希夫碱配体中的酚羟基氧原子O3、亚氨基氮原子N1、羧基氧原子O1、2,2′-联吡啶配体上的2个氮原子N4和N5以及咪唑环上的氮原子N2i配位,形成一个六配位的变形八面体结构N1,N4,O1和O3原子位于八面体的赤道平面上N2i和N5原子位于八面体的轴向位置。赤道平面上的键角∠N1-Ni1-O3(91.7(2)°),∠N1-Ni1-O1(80.7(2)°)∠O3-Ni1-N4(92.5(2)°)和∠O1-Ni1-N4(95.1(2)°)之和为360.0°,表明Ni(Ⅱ)离子几乎与赤道平面共面,其偏离基本平面位置距离为0.008 19(3)nm,N2i-Ni1 N5形成的键角为170.2(3)°,表明配位原子围绕Ni(Ⅱ)离子形成一畸变的八面体。另外联吡啶平面分子与Ni(Ⅱ)离子配位形成一个五元环,其基本平面与赤道平面之间形成的二面角为87.02(18)°,几乎垂直于赤道平面。希夫碱配体围绕Ni(Ⅱ)离子形成Ni1 O1-C1-C2-N1五元环和 Ni1-O3-C9-C8-C7-N1六元环,两环基本平面所形成的二面角为9.20(31)°,它们的形成增加了配合物的稳定性。在配合物2的晶体中,溶剂甲醇分子通过氢键O4-H4…O3iii与主体分子连接。溶剂水分子通过N3-H3…O5ii、O5-H5D …O2v、O6-H6D…O2vii及O6-H6C…O2vi等与主体分子形成氢键作用。另外,2个溶剂水分子之间通过O5-H5C…O6iv形成氢键作用。这些氢键相互作用将配合物分子连接形成二维平面网状结构(图2),其键长和键角见表3。
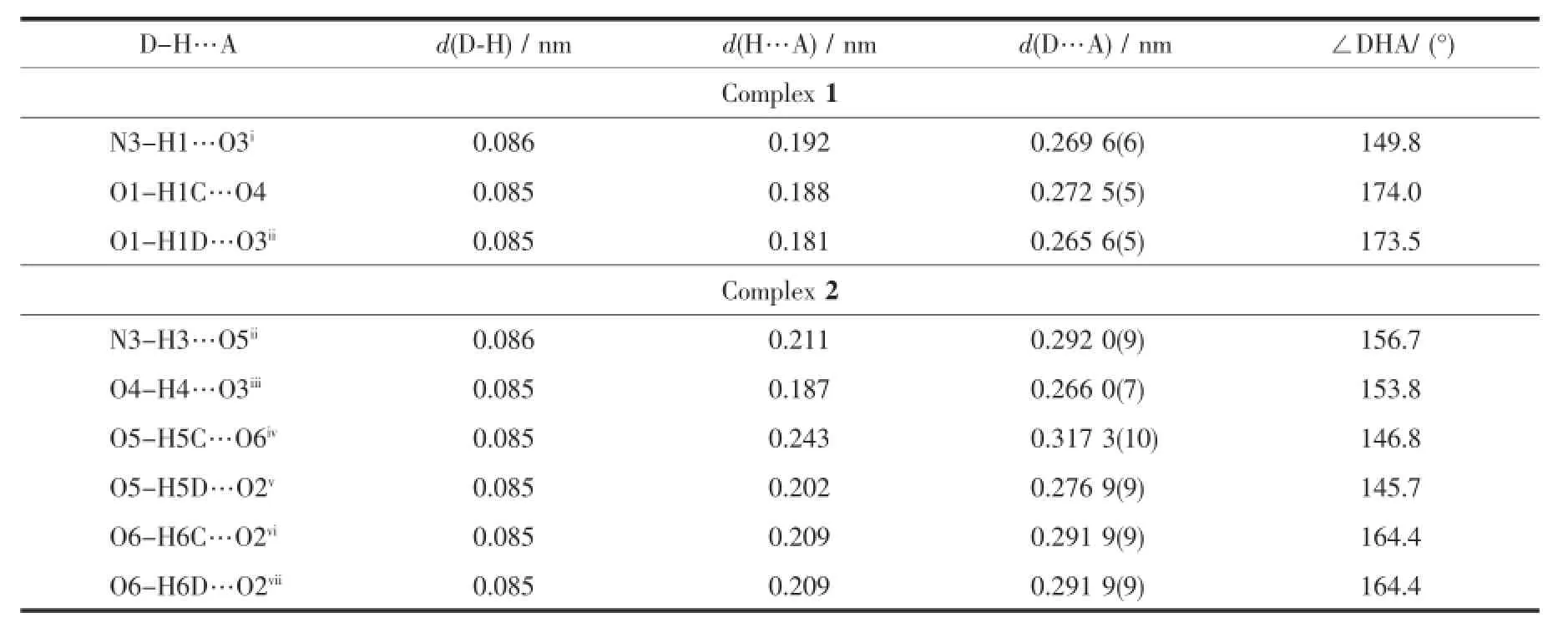
表3 配合物1和2的氢键键长和键角Table 3 Hydrogen bond lengths(nm)and angles(°)for complex 1 and 2
2.2配合物与DNA作用
2.2.1紫外可见吸收光谱
电子吸收光谱法是研究配合物与DNA相互作用的常用方法。当金属配合物与DNA结合后,其配体所处的环境会发生变化,从而导致配合物的吸收光谱的强度和波长发生变化。一般来说,若金属配合物与DNA之间的相互作用为静电或沟面作用方式时,它们的紫外光谱不会发生明显的变化。但加入DNA后,若配合物的电子吸收光谱的吸收峰位红移,强度减小,可认为是配合物与DNA发生插入作用的证据之一[19]。因为插入配体与DNA碱基对可发生π电子堆积,插入配体的π*空轨道与碱基的π轨道发生偶合,使能量降低,导致π→π*跃迁能量减小,从而产生红移现象;同时,偶合后的π*轨道因部分填充电子使π→π*跃迁几率减小,产生减色效应[20]。
图3为不同浓度CT-DNA存在下配合物的紫外吸收光谱图。从图中可以看出,随着DNA浓度的增大,配合物1在268 nm处的吸收发生了明显的减色效应且稍有红移,这说明配合物与DNA的作用方式是插入作用。同样配合物2也产生了相似现象。为了定量衡量配合物与DNA结合的强弱,可利用公式cDNA/(εa-εf)=cDNA/(εb-εf)+1/[Kb(εb-εf)]求得结合常数Kb[21],其中εa为配合物的表观摩尔吸光系数,εb,εf分别为键合和自由配合物的摩尔吸光系数。将-cDNA/(εa-εf)对cDNA作图(图3),求得结合常数Kb(1)=2.41×104L·mol-1,Kb(2)=1.89×104L·mol-1。作为与DNA结合的典型的插入试剂EB,它与DNA的结合常数为3.3×105L·mol-1[22],与之相比,这两个配合物比EB插入剂的结合常数小,这说明该配合物与DNA的插入作用较弱,这可能是因为配合物为畸变的八面体构型,其平面性没有EB的好。
2.2.2荧光光谱
溴化乙锭(EB)是研究药物分子与DNA作用的经典的嵌入式荧光探针,它自身的荧光很弱,但是与DNA作用后,其生色团嵌入DNA分子的碱基对中使荧光强度大大增加[23]。当有其它也能与DNA发生插入作用的配合物存在时,会将EB从EB-DNA复合物中挤出,从而使EB-DNA的荧光强度显著降低。因此,通过测定体系荧光的降低程度,可以推断配合物与DNA的相互作用方式。
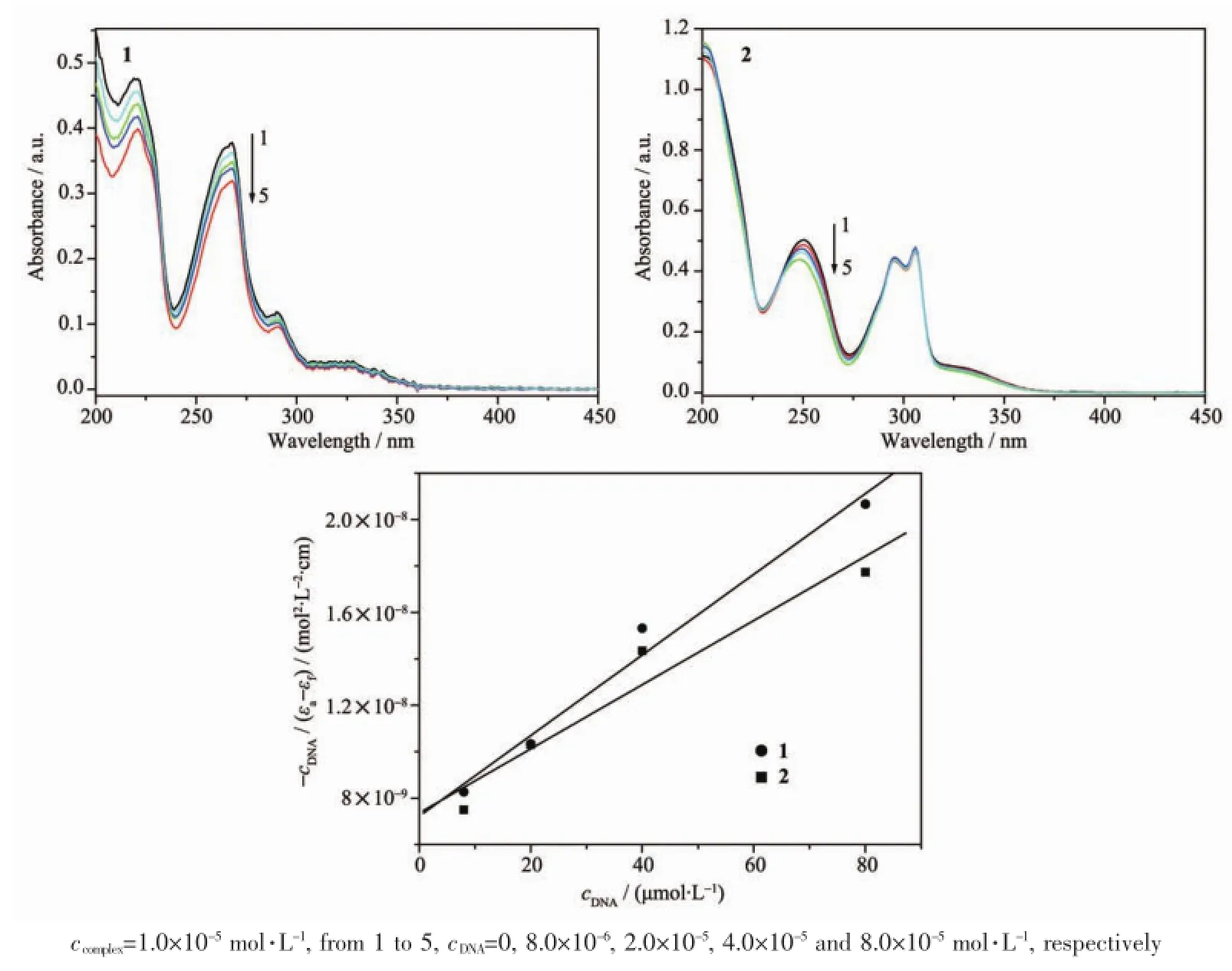
图3 不同浓度CT-DNA存在下配合物1和2的紫外可见吸收光谱以及-cDNA/(εa-εf)对cDNA作图Fig.3 Absorption spectra of complex 1 and 2 in the absence and presence of CT-DNA,and the plot of-cDNA/(εa-εf)vs cDNA
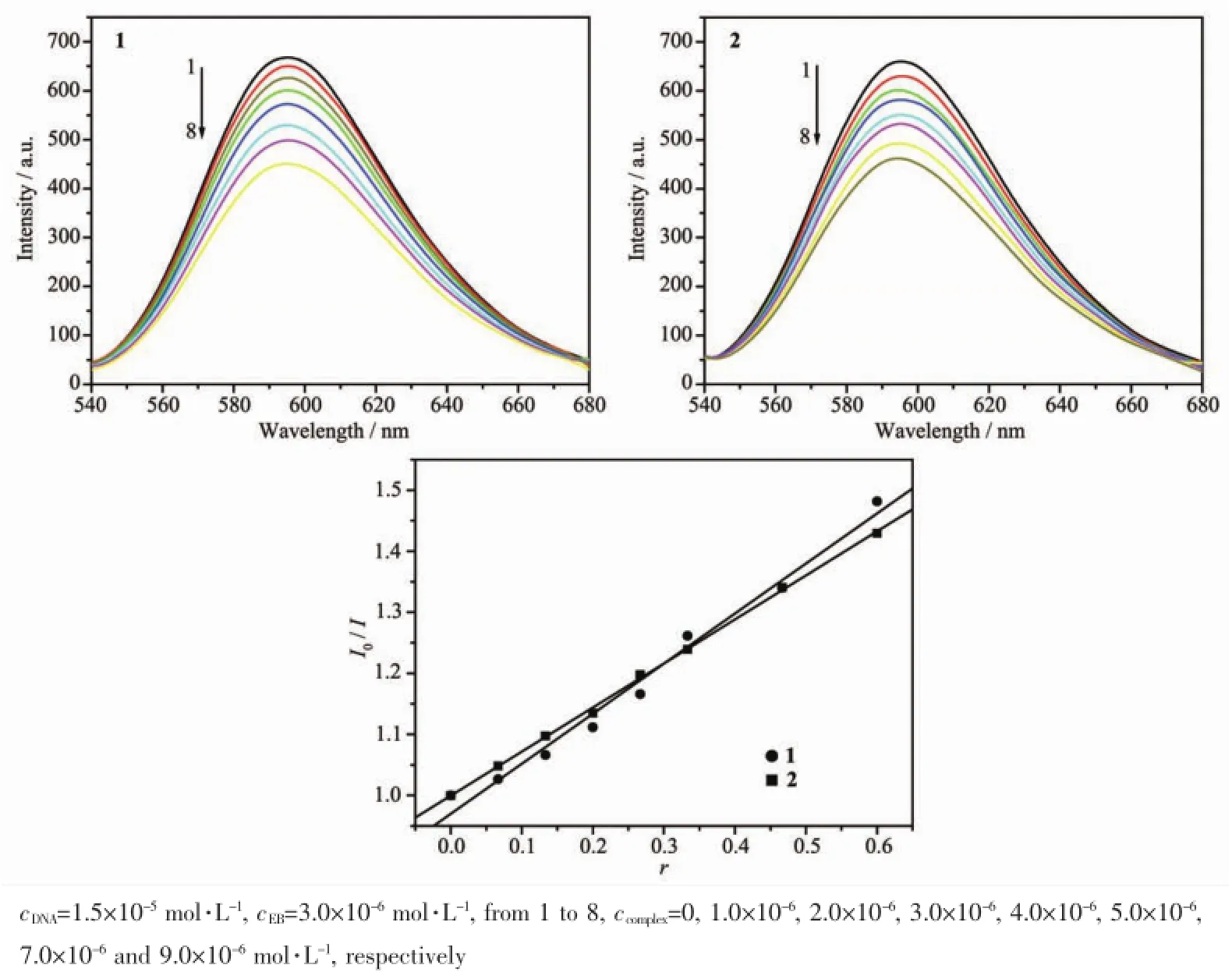
图4 不同浓度配合物存在时EB-DNA体系的荧光光谱和I0/I对r作图Fig.4 Fluorescence spectra of EB-DNA system in the absence and presence of complex 1 and 2,and plot of I0/I vs r
图4为配合物与EB竞争实验的荧光光谱图。由图4可见,EB-DNA体系在595 nm处的荧光强度随着2个配合物浓度的增加而显著降低,这说明配合物发生了类似于EB的插入作用,从而将EB从结合位点竞争下来。为定量研究这2个配合物对EB-DNA体系的荧光猝灭程度,可根据Stern-Volmer方程求得荧光猝灭常数Ksq[24]:I0/I=1+Ksqr,其中I0为未加入配合物时EB-DNA体系的荧光强度,I为加入不同浓度配合物时EB-DNA体系的荧光强度,r=ccomplex/cDNA。将I0/I对r作图,分别求得猝灭常数Ksq(1)=0.85,Ksq(2)=0.72。由此看出,配合物1的作用要强于配合物2,这可能是菲咯啉的平面性比2,2′-联吡啶的要好,可以更好地嵌入到DNA的碱基对中。这与吸收光谱的实验结果一致。与所报道的镍配合物[Ni(PzTA)2CO3]·5H2O(Ksq=0.542),[NiQ(PyTA)(H2O)2]Cl·H2O(Ksq=0.708)和[NiQ(PzTA)(H2O)2]Cl·H2O(Ksq= 0.613)[25]相比要大些,但是明显小于[Ni(atz)(DCA)(H2O)2]·2H2O(Ksq=1.38)[26],与这些配合物的结构不同有关。
2.2.3圆二色光谱
CD光谱是研究小分子与DNA相互作用并探究其对DNA构象影响的重要手段之一。CT-DNA的CD光谱在273 nm和245 nm处有2个明显的特征峰,273 nm处正峰是由CT-DNA碱基对的π-π堆积作用引起的,245 nm处负峰则主要对应于双螺旋结构的B型构象[27]。图5为配合物与CT-DNA作用的圆二色光谱。由图中可以看出,当加入相同浓度的配合物1和2后,DNA的CD谱发生了明显的变化,273 nm处正峰逐渐增大,而245 nm处负峰基本不变,且配合物1引起的正峰增大值比配合物2更强。这说明2个配合物与DNA的作用是插入到DNA碱基对中,影响到DNA碱基对之间的π-π堆积作用使DNA的构象发生了改变[28],并且配合物1对其构象的影响要强于配合物2。这与紫外可见吸收光谱和荧光光谱的结论是吻合的。
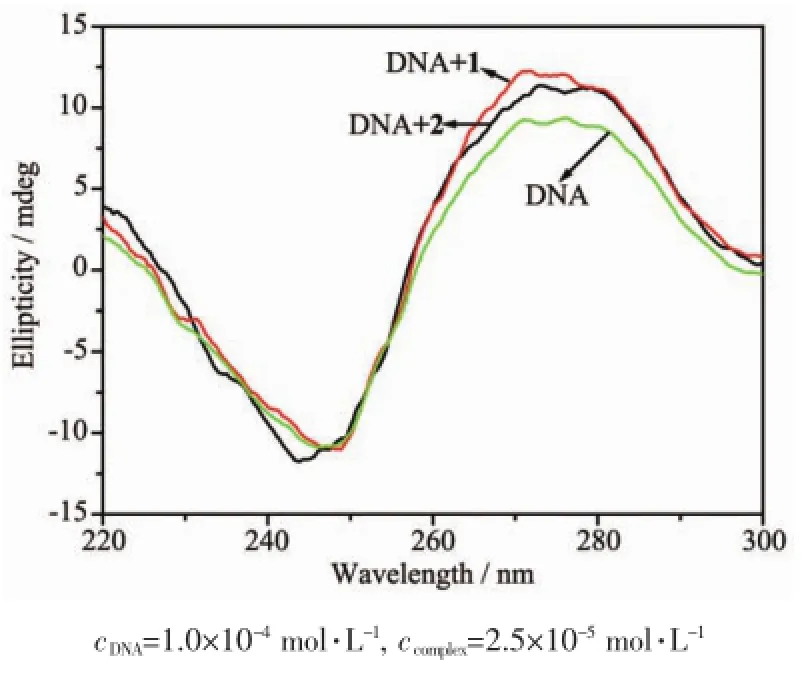
图5 配合物对CT-DNA圆二色光谱的影响Fig.5 Effects of the two complexes on CD spectra of CT-DNA
2.2.4粘度测定
粘度测定被认为是在缺乏晶体结构数据的情况下,确定配合物与DNA结合模式最有效的方法之一,其结果有时比光谱数据更具有说服力[29]。配合物对DNA溶液粘度的影响有3种情况:(1)当配合物以沟面或静电方式与DNA作用时,对DNA双链的影响较小,DNA溶液的粘度几乎不发生改变;(2)当配合物以部分插入方式 (或沟槽结合)进入DNA碱基对时,使得DNA双链发生扭结,降低DNA溶液的粘度;(3)当配合物以插入方式与DNA作用时,DNA相邻碱基对的距离会变大来容纳配合物小分子的插入,因而导致DNA双螺旋链伸长,使DNA溶液的粘度增大。图6为不同化合物对DNA溶液粘度的影响。由图6可看出,随着配合物浓度的增加,DNA溶液的相对粘度增大,与插入剂EB所引起DNA溶液的粘度变化趋势一致,而与沟槽结合剂甲基绿(MG)的变化趋势相反。这说明配合物1和2均以插入方式与CT-DNA结合,但比典型的插入剂EB要弱;且配合物1与DNA的插入作用要略强于配合物2。这与上述光谱研究结果是一致的。这可能是由于配合物1中配体phen的平面性好,且分子体积较小而空间位阻较小,有利于其与DNA的插入作用。
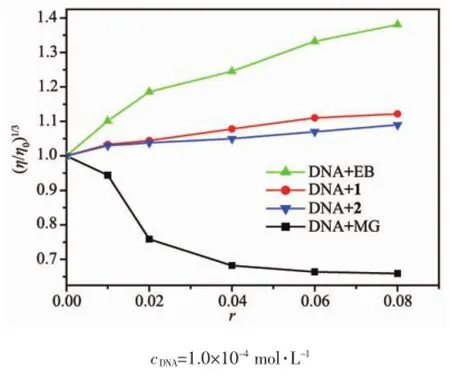
图6 配合物、EB和MG对CT-DNA相对粘度的影响Fig.6 Effects of the complexes,EB and MG on the relative viscosity of CT-DNA
2.3SOD活性测定
氮蓝四唑(NBT)光照还原法是测定物质SOD活性的常用的方法。在光照条件下,核黄素(VB2)与四甲基乙二胺反应产生超氧阴离子自由基(O2-·),生成的O2-·使NBT还原为蓝紫色化合物甲臜,甲臜在560 nm处有最大吸收,且吸光度与浓度成正比,因此通过测定不同时间下吸光度A560的变化可以得到一条直线,直线斜率(K值)的大小可以反映甲臜生成的快慢。当向该体系中加入SOD活性配合物后,其与NBT竞争O2-·,从而减慢甲臜生成的速率。图7为不同浓度的配合物对反应体系吸光度随时间变化曲线。由图7可知,随着配合物浓度的增大,直线的斜率逐渐变小,说明浓度越大,抑制率越大。配合物对O2-·的抑制率(α)可以通过公式α=(1-k′/k)×100%求出[30],其中k为未加配合物之前的直线斜率,k′为加入配合物后的直线斜率。以抑制率对配合物浓度作图,可获得抑制率为50%时的配合物浓度,即为一个活性单位IC50。IC50越小,活性越大。图8为以抑制率α对配合物浓度作图所得曲线,求得配合物1的IC50=51.5 μmol·L-1,配合物2的IC50= 58.1 μmol·L-1。其值与之前报道的镍配合物[Ni(L)(HL)](ClO4)·H2O(IC50=34.0 μmol·L-1)和[Ni(HL)(bipy)(H2O)](NO3)(ClO4)·H2O(IC50=38.0 μmol·L-1)的IC50值相当[31],但与天然SOD(IC50=0.015 μmol·L-1)相比,相差较大[32]。这说明该配合物具有一定的SOD活性。
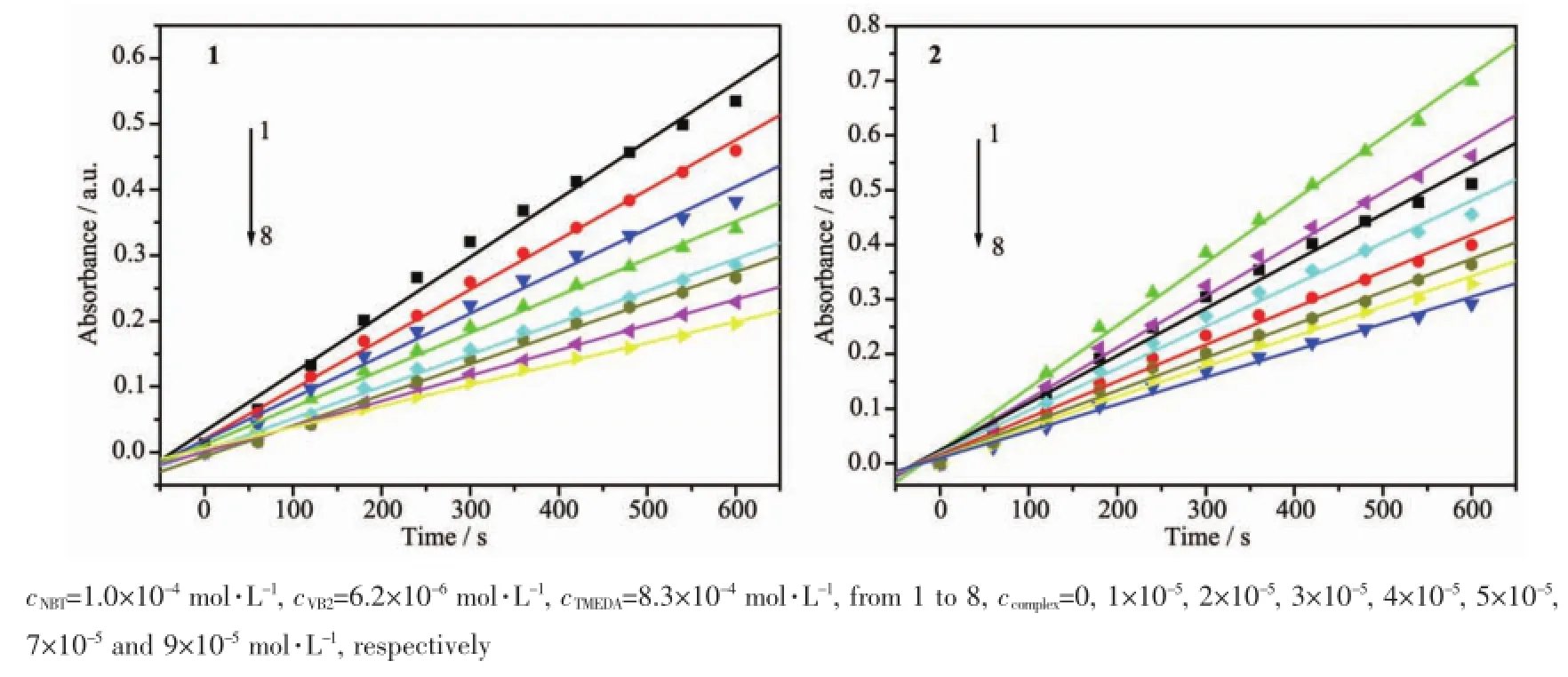
图7 配合物的浓度对反应体系的吸光度(A560)-时间关系的影响Fig.7 Effect of complex concentration on the plot of absorbance of the reaction system vs time
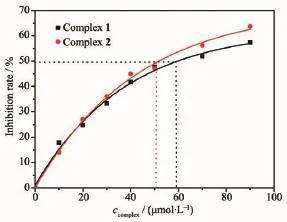
图8 配合物对O2-·的抑制率随浓度变化曲线Fig.8 Change of inhibition rate of the complex to O2-· with various concentrations
3 结论
合成了L-组氨酸水杨醛希夫碱与菲咯啉或2,2′-联吡啶混配体的2个镍(Ⅱ)配合物,通过IR进行了表征,利用X射线单晶衍射测定了其晶体结构。利用紫外可见吸收光谱、荧光光谱、圆二色光谱和粘度测定等研究了配合物与CT-DNA的相互作用,结果表明配合物均以插入方式与CT-DNA作用,而配合物1对DNA的作用要强于配合物2,这是由于菲咯啉的平面性比2,2′-联吡啶的要好,可以更好的插入到DNA的碱基对中。NBT光照还原法测定表明2种配合物都具有一定的SOD活性。这些结果可为研究氨基酸希夫碱镍配合物的结构和生物活性及作用机理提供一定的实验证据。
参考文献:
[1]Alexiou M,Tsivikas I,Dendrinou-Samara C,et al.J.Inorg Biochem.,2003,93(3):256-264
[2]Abdel-Rahman L H,El-Khatib R M,Nassr L A E,et al.J Mol.Struct.,2013,1040:9-18
[3]Ainscough E W,Dobbs A J,Ranford J D,et al.Inorg.Chim Acta,1998,267(1):27-38
[4]Boer J L,Mulrooney S B,Hausinger R P,et al.Arch.Biochem Biophys.,2014,544:142-152
[5]Nitha L P,Aswathy R,Mathews N E,et al.Spectrochim.Act Part A,2014,118:154-161
[6]Selvakumaran N,Bhuvanesh N S P,Endo A,et al.Polyhedron 2014,75:95-109
[7]Jung Y,Lippard S J.Chem.Rev.,2007,107:1387-1407
[8]Krishnamoorthy P,Sathyadevi P,Butorac R R,et al.DaltoTrans.,2012,41:4423-4436
[9]Sawyer D T,Valentine J S.Acc.Chem.Res.,1981,14:393-400
[10]TAN Shi-Dong(覃事栋),FENG Si-Si(冯思思),ZHANG Hong -Mei(张红梅),et al.Acta Chim.Sinica(化学学报),2005,63: 1155-1160
[11]GAO Yue-Ying(高月英),XU Ke-Wei(徐克伟),MAO Yue-Wa(毛岳洼),et al.Acta Chim.Sinica(化学学报),2001,59: 1176-1183
[12]Reichmann M E,Rice S A,Thomas C A,et al.J.Am.Chem. Soc.,1954,76(11):3047-3053
[13]Sheldrick G M.SAINT Ver.4,Software Reference Manual,Siemens Analytical X-ray Systems,Madison,WI,1996.
[14]Sheldrick G M.SADABS,Program for Empirical Absorption Correction of Area Detector Data,University of Göttingen,Germany,1996.
[15]Sheldrick G M.SHELXTL 6.10,Bruker Analytical Instrumentation,Madison,Wisconsin,USA,2000.
[16]DING Yang(丁杨),DENG Jian-Sheng(邓剑生),LE Xue-Yi(乐学义).Chinese J.Org.Chem.(有机化学),2011,31(7): 1081-1086
[17]BIAN Lin(边琳),LI Lian-Zhi(李连之),WANG Xia(王霞),et al.Chinese J.Inorg.Chem.(无机化学学报),2011,27: 649-654
[18]PU Xue-Wei(蒲学炜),LI Lian-Zhi(李连之),DONG Jian-Fang(董建方),et al.Acta Chim.Sinica(化学学报),2011,69:647-652
[19]Tysoe S A,Baker A D,Strekas T C.J.Phys.Chem.,1993,97 (8):1707-1711
[20]LI Hong(李红),YUE Xue-Yi(乐学义),WU Jian-Zhong(吴建中),et al.Acta Chim.Sinica(化学学报),2003,61(2):245-250
[21]Wolfe A,Shimer G H,Meehan T.Biochemistry,1987,26 (20):6392-6396
[22]Strothkamp K G,Strothkamp R E.J.Chem.Educ.,1994,71 (1):77-79
[23]WU Jian-zhong(吴建中),WANG Lei(王雷),YANG Guang(杨光),et al.Chem.J.Chinese Universities(高等学校化学学报),1996,17(7):1010-1015
[24]Lakowicz J R,Weber G.Biochemistry,1973,12(21):4161-4170
[25]Duan R R,Ou Z B,Wang W,et al.Spectrochim.Acta Part A,2015,151:64-71
[26]Wang N,Lin Q Y,Wen Y H,et al.Inorg.Chim.Acta,2012,384(1):345-351
[27]Uma V,Kanthimathi M,Weyhermuller T,et al.J.Inorg. Biochem.,2005,99(12):2299-2307
[28]Lincoln P,Tuite E,Norden B.J.Am.Chem.Soc.,1997,119 (6):1454-1455
[29]Qiu B,Guo L H,Wang W,et a1.Biosens.Bioelectron.,2007,22(11):2629-2635
[30]Le X,Liao S,Liu X,et al.J.Coord.Chem.,2006,59(9):985-995
[31]Patel R N,Singh A,Sondhiya V P,et al.J.Coord.Chem.,2012,65(5):795-812
[32]DING Yang(丁杨),SHEN Shu-Yi(沈淑仪),REN Xiang-Xiang(任祥祥),et al.Chemistry(化学通报),2009,72(10): 922-926
中图分类号:O614.81+3
文献标识码:A
文章编号:1001-4861(2016)05-0789-10
DOI:10.11862/CJIC.2016.117
收稿日期:2015-12-12。收修改稿日期:2016-03-24。
Syntheses,Crystal Structures,DNA Interactions and SOD Activities of Two Nickel(Ⅱ)Complexes with L-Histidine Schiff Base
WEI Qiang1DONG Jian-Fang2LI Wen-Bin1ZHAO Pei-Ran1DING Fei-Fei1LI Lian-Zhi*,1
(1School of Chemistry and Chemical Engineering,Liaocheng University,Liaocheng,Shandong 252059,China)
(2Department of Material Science,Shandong Polytechnic Technician College,Liaocheng,Shandong 252027,China)
Abstract:Two new nickel(Ⅱ)complexes,[Ni(Sal-L-His)(Phen)]·H2O(1)(Sal-L-His=the Schiff base derived from salicylaldehyde and L-Histidine,Phen=1,10-phenanthroline),and[Ni(Sal-L-His)(Bipy)]2·2CH3OH·3H2O(2)(Bipy= 2,2′-Bipyridine),have been synthesized.Their crystal structures have been determined by single crystal X-ra diffraction analysis.It showed that 1 is a mononuclear complex and belongs to monoclinic crystal system,P21/ space group with the cell parameters:a=1.586 31(13)nm,b=1.215 11(11)nm,c=1.211 30(9)nm,β=106.581(2)° V=2.237 7(3)nm3,Z=4;2 is a binuclear complex and belongs to orthorhombic crystal system,C2221space group with the cell parameters:a=1.415 30(13)nm,b=1.717 71(15)nm,c=2.256 02(19)nm,V=5.484 6(8)nm3,Z=4 The results of UV absorption,fluorescence,circular dichroism(CD)spectra and viscosity measurement indicated that the two complexes bind to CT-DNA in an same intercalative mode.SOD-like activities of the complexes were determined by the modified photoreduction of NBT.The value of IC50(1)is 51.5 μmol·L-1and IC50(2)is 58.1 μmol· L-1.CCDC:1038774,1;1420088,2.
Keywords:nickel(Ⅱ)complex;L-histidine Schiff base;crystal structure;DNA;SOD activity
猜你喜欢
食品与发酵工业(2021年12期)2021-07-05 14:38:46
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
中国蔬菜(2019年4期)2019-06-06 08:08:56
中国饲料(2018年7期)2018-01-24 03:38:16
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
饲料博览(2016年7期)2016-04-05 14:20:34
火炸药学报(2014年3期)2014-03-20 13:17:39
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
