
基于mtDNA Cytb基因序列的我国北方地区甜菜夜蛾遗传多样性与种群历史分析
2016-07-21 01:25王兴亚周俐宏
生态学报 2016年8期
王兴亚,周俐宏
1 辽宁省农业科学院植物保护研究所,沈阳 110161 2 辽宁省农业科学院花卉研究所,沈阳 110161
基于mtDNACytb基因序列的我国北方地区甜菜夜蛾遗传多样性与种群历史分析
王兴亚1,*,周俐宏2
1 辽宁省农业科学院植物保护研究所,沈阳110161 2 辽宁省农业科学院花卉研究所,沈阳110161
摘要:为了明确我国北方不同地理种群甜菜夜蛾Spodoptera exigua遗传多样性与种群遗传结构,阐明该种害虫的种群历史动态,首次对采自我国北方8省17县(市)304头甜菜夜蛾样品进行mtDNA Cytb基因序列测定与分析,利用DnaSP 5.0和Arlequin 3.0软件分析种群遗传多样性、遗传结构、遗传分化与分子变异,基于MP、ML与贝叶斯法构建单倍型系统发育树,与此同时,基于Median-joining法对所有个体构建单倍型网络关系图。结果表明,在所分析的304个序列样本中,共检测出19个单倍型,其中,包括9个共享单倍型, 单倍型Hap6为所有种群所共享。总群体具有较低的遗传多样性(Hd=0.422±0.035,π=0.00119±0.00011)与较小的遗传分化(FST=0.108,P<0.001)。单倍型系统发育分析与网络关系图结果表明,虽然19个单倍型被分为2个分支,但各单倍型相互散布在不同种群中,未形成明显谱系地理格局。AMOVA分析表明,甜菜夜蛾遗传变异主要来自种群内(89.18%),种群间变异水平较低(10.82%)。中性检验(Tajima′s D = -1.897, P<0.05; Fu′s FS = -4.424, P<0.05)与错配分布分析表明,我国北方地区甜菜夜蛾种群曾经历过种群的近期扩张。
关键词:甜菜夜蛾;细胞色素b(Cytb);线粒体DNA;种群历史;遗传分化
甜菜夜蛾Spodopteraexigua(Hübner)属鳞翅目Lepidoptera夜蛾科Noctuidae灰夜蛾属Spodoptera,是我国一种重要杂食性害虫,可危害玉米、大豆、棉花、蔬菜等多种作物,造成严重的经济损失[1- 3]。该种害虫起源于南亚,广泛分布于亚洲、欧洲、非洲及北美洲等热带与温带地区[4]。19世纪90年代,我国甜菜夜蛾首次在北京有发生与危害记录。近年来,随着全球气候转暖,甜菜夜蛾已迅速扩散到我国北方蔬菜产区,成为危害大葱等蔬菜的最重要害虫。通常,该种害虫幼虫取食寄主叶片,导致作物减产,如不及时防治,最终可造成寄主死亡[5- 6]。另外,甜菜夜蛾亦是一种高繁殖率、具有长距离迁移能力的害虫[7- 9]。尤其是,近年来由于长期大量的使用杀虫剂,使得甜菜夜蛾对有机磷类、氨基甲酸酯等杀虫剂产生严重抗药性,给该种害虫的防治带来更大困难[10- 11]。
近年来,许多分子标记被广泛应用于物种系统发育和生物地理学等研究方面[12- 13]。由于线粒体基因(mtDNA)具有严格的母系遗传、缺少基因重组以及较快的进化速率等特点,而被广泛用于研究种群进化历史、谱系地理学及物种形成等方面[13- 17]。其中,细胞色素b(cytochrome b,Cytb)基因是目前昆虫线粒体基因组13个编码蛋白质基因中结构与功能研究最为清楚的基因[18- 19]。并且,由于该基因进化速率适中,适合昆虫种及种下阶元的分类鉴定以及群体遗传变异[20- 24]。迄今,国内外在DNA水平上探讨甜菜夜蛾的种群遗传学研究甚少。张艳研究了甜菜夜蛾抗性遗传及AFLP体系的建立[25];孙小洁等建立了甜菜夜蛾cDNA表达文库[26];牛成伟等利用AFLP研究了我国北方甜菜夜蛾种群内遗传多样性高于南方种群,不同种群间不存在明显遗传分化[27]。
鉴于此,本研究利用mtDNACytb基因序列作为分子标记,对我国北方不同地理种群甜菜夜蛾的种群遗传多样性、遗传结构、遗传分化及种群历史进行深入分析,为阐明该种害虫在我国北方的种群历史动态,进而为制定合理的害虫防治策略提供理论依据。
1材料与方法
1.1实验材料与采集方法
本研究选取2012年采自我国8省17县(市)的304头甜菜夜蛾样品,该样品采用实地采集幼虫与性信息素诱捕雄性成虫方法获得(表1)。为了尽量避免采集来自同一个父母本的后代个体,采集时尽量保持一定距离。采集到的所有样品均浸泡在- 20 ℃,95%乙醇中,并保存于辽宁省农业科学院植物保护研究所(中国,沈阳)。另外,在构建系统发育树时,采用Spodopteraandrogea(Stoll)、西部黄条粘虫夜蛾Spodopterapraefica(Grote)和Spodopteralatifascia(Walker)作为外群。

表1 我国北方地区不同地理种群甜菜夜蛾样品的采集信息
1.2基因组总DNA的提取、PCR扩增与测序
利用Qiagen′s DNEasy提取试剂盒(Qiagen,Valencia,CA)进行甜菜夜蛾总DNA提取,选用mtDNACytb部分序列作为分子标记,其扩增引物为CP1(5′-GATGATGAAATTTTGGATC-3′)[28]和TRs(5′-TATTTCTTTATTATGTTTTCAAAAC-3′)[29]。PCR反应均为25 μL反应体系,包含0.25 μL EasyTaq DNA Polymerase(5U/μL);2.5 μL 10×Easy Taq Buffer(+Mg2+);0.5 μL dNTP (2.5 mmol/L);正反引物各0.5 μL (10 μmol/L);1 μL DNA模板;19.75 μL灭菌水。35个PCR循环参数为:94 ℃预变性5 min;94 ℃变性30 s,46 ℃退火30 s,72 ℃延伸45 s,30个循环;72 ℃终延伸5 min。PCR产物在4 ℃保存。PCR反应在Applied Biosystems ABI 3730(Applied Biosystem,USA)上进行。所得双向测序结果通过DNAStar 5.0(DNASTAR,Inc. 1996)软件包中的Seqman程序进行校对及双向测序结果的拼接。
1.3序列处理与数据分析
运用Clustal X 1.81[30]软件进行多重序列比对,使用MEGA 5.2[31]软件基于Kimura双参模型(Kimura 2-parameter)计算全部碱基替换情形下的序列间遗传距离(K2P-distance)[32]。计算公式:
式中,P和Q分别代表转换和颠换类型的同源位点概率值。
利用最大简约法(MP)、最大似然法(ML)和贝叶斯法(BI)构建系统发育树,估计单倍型之间的系统发育关系。MP树在PAUP*4.10b[33]上进行,采用启发式搜索,随机加样,重复1000次,构建最大简约树。用Bootstrapping分析,对各分支点进行评价,检测1000次,每次检测随机加样重复100次。获得的50%合意树,如超过1个最大简约树,利用Modeltest 3.7[34]和Akaike information criterion(AIC)[35]进行最适进化模型估算。ML树也在PAUP*4.10b上执行,采用启发式搜索,随机加样,重复100次,构建最大似然树。用bootstrapping分析,对各分支点进行评价,检测100次,每次检验随机加样重复10次。BI系统树分析,以随机树为起始树,根据其最适模型,4条马尔可夫链运行1000000代,每200代抽样并保存1次数据,保存树的分支长度。抛弃前2500代,将抽样得到的前500棵树舍弃,以保证树的似然率-LnL达到最大并趋于稳定。
使用NETWORK 2.0[36]进行基于Median-joining法对所有个体构建单倍型之间的网络关系图。使用DnaSP 5.0统计单倍型多态度(Hd)、核苷酸多态度(π),以及分子标记的变异速率的中性检验[37]。应用Arlequin 3.0[38]进行分子变异分析(analysis of molecular variance, AMOVA)与基于Kimura 2-parameter遗传距离的种群遗传分化FST值统计,FST值计算公式如下[39]:
式中,ni代表在第i种群样本中的基因拷贝数。
2结果与分析
2.1Cytb序列变异及遗传多样性
通过实验得到8省17个地理种群的304头甜菜夜蛾的测序结果,Cytb基因序列长度为798 bp。其中,776个碱基序列为保守位点,22个变异位点(占序列总长度的2.8%),9个简约信息位点。没有出现碱基插入和缺失现象。平均碱基组成分别为42.7% A,33.1% T,12.6% G和11.5% C,A+T含量(75.8%)明显高于G+C含量(24.2%),严重A+T偏向性与其它鳞翅目昆虫相一致[40- 41]。
由表2可以看出,17个地理种群甜菜夜蛾的总群体单倍性多态度(Hd)为0.422 ± 0.035,核苷酸多态度(π)为0.00119 ± 0.00011。其中,BZ种群的单倍型多态度(Hd)和核苷酸多态度(π)最高,分别为0.643 ± 0.106和0.00214 ± 0.00039。与之相比较,SY和HAD种群的单倍型多态度(Hd)和核苷酸多态度(π)最低,皆为0。
2.2单倍型分布与系统发育分析
在304头甜菜夜蛾Cytb基因序列中,共检测出19个单倍型(GenBank登录号:KF589828-KF589846)。各地理种群单倍型分布见表2。各地理种群单倍型数量范围为1—5个,平均具有单倍型3.11个,其中,DL、YANC、BZ和PENL种群单倍型数量最丰富,分别在26、12、19、20个检测个体中皆共定义了5个单倍型。在此19个单倍型中,包括9个共享单倍型(Hap1、Hap3、Hap5、Hap6、Hap8、Hap12、Hap14、Hap16和Hap17)。其中,Hap1广泛分布于所有5个种群的14个样本中;Hap6广泛分布于所有全部17个种群的228个样本中;Hap14广泛分布于所有12个种群的31个样本中。另外,还包括10个独有单倍型,其中,Hap2、Hap4、Hap7、Hap13、Hap15、Hap18和Hap19分别为LUC、DL、XINX、TA、BZ、YANC和SY种群所特有,Hap9、Hap10、Hap11为PENL种群所特有。
以S.androgea、S.praefica和S.latifascia作为外群,基于最大简约树(MP)、最大似然树(ML)与贝叶斯树(BI)构建单倍型系统发育树(图1),结果表明,尽管MP/ML系统树的后验概率较低,系统发育树被分为2个分支(Clade),但也存在其他尚未解决的分支。Clade I中包括Hap6—Hap12,Clade II中包括Hap13—Hap19。与此同时,基于Median-joining法构建的单倍型网络关系的研究结果表明,单倍型Hap6广泛分布于全部17个地理种群中。单倍型之间通常仅由1—2个突变所联系,其中,Hap6分别与Hap7、Hap8、Hap9、Hap10和Hap13之间仅有1个突变联系(图2)。该单倍型网络关系与系统发育结果一致,各单倍型都相互散布在不同的地理种群中,未形成明显的系统地理格局。
表2我国北方不同地理种群甜菜夜蛾的单倍型分布、遗传多样性及中性检测
Table 2Distribution of the haplotypes, genetic diversity and neutral test among different geographic populations ofSpodopteraexiguain North China

种群Population单倍型数NumberofHaplotype(n)单倍型(个体数)Haplotypes(numberofindividuals)单倍型多态度Haplotypediversity(Hd±SD)核苷酸多态度Nucleotidediversity(π±SD)中性检验及显著性检验NeutralitytestandsignificancetestTajima'sD值Tajima'sDvalueFu'sFS值Fu'sFSvalueSY4Hap5(1),Hap6(9),Hap14(1),Hap19(1)0.455±0.1700.00173±0.000700.0000.000FX3Hap1(3),Hap6(14),Hap14(2)0.444±0.1240.00111±0.000310.09680.905HLD4Hap5(1),Hap6(20),Hap8(5),Hap14(1)0.430±0.1030.00085±0.00030-1.339-0.607DAL2Hap6(12),Hap14(4)0.400±0.1140.00100±0.000280.8382.177TJ2Hap6(6),Hap16(2)0.429±0.1690.00161±0.000630.4582.469XINL2Hap6(10),Hap14(1)0.182±0.1440.00046±0.00036-1.430*0.506HAD1Hap6(17)0.000±0.0000.00000±0.000000.0000.000LUC4Hap1(5),Hap2(1),Hap6(1),Hap14(4)0.709±0.0990.00201±0.00040-0.2400.266YC3Hap6(13),Hap14(3),Hap17(1)0.404±0.1300.00111±0.00036-0.0160.784DL5Hap1(4),Hap3(2),Hap4(1),Hap6(16),Hap14(3)0.600±0.0980.00176±0.000310.2010.098NAY2Hap5(1),Hap6(16)0.118±0.1010.00044±0.00038-1.706*0.708XINX3Hap1(1),Hap6(20),Hap7(1)0.177±0.1060.00034±0.00022-0.8350.813YANC5Hap1(1),Hap6(8),Hap14(1),Hap17(1),Hap18(1)0.576±0.1630.00213±0.00070-0.556-0.609TA3Hap6(26),Hap12(3),Hap13(1)0.246±0.0980.00055±0.00023-0.989-0.190ZQ2Hap6(17),Hap14(3)0.268±0.1130.00067±0.00028-0.1121.524BZ5Hap3(3),Hap5(1),Hap6(11),Hap14(3),Hap15(1)0.643±0.1060.00214±0.00039-0.0170.183PENL5Hap6(12),Hap9(1),Hap10(1),Hap11(1),Hap14(5)0.600±0.1010.00149±0.00032-0.945-0.672总计Total19Hap1(14),Hap2(1),Hap3(5),Hap4(1),Hap5(4),Hap6(228),Hap7(1),Hap8(5),Hap9(1),Hap10(1),Hap11(1),Hap12(3),Hap13(1),Hap14(31),Hap15(1),Hap16(2),Hap17(2),Hap18(1),Hap19(1)0.422±0.0350.00119±0.00011-1.897*-4.424*
加粗单倍型为独有单倍型,SD表示标准差,*表示差异显著(P<0. 05)

图1 基于mtDNA Cytb基因的单倍型序列的甜菜夜蛾系统发育分析Fig.1 Phylogenetic trees of Spodoptera exigua based on haplotype sequence variation of partial mtDNA Cytb sequences MP/ML分析的自举检验与BI分析的后验概率分析表示在枝的上部 [MP树:一致性参数 (Consistent index, CI),0.862;保留指数 (Retention index, RI),0.840;校正一致性指数 (Rescaled consistency, RC),0.725;树长,138;GTR+I为ML和Bayesian分析的最优模型];Spodoptera androgea (HQ177620)、Spodoptera praefica (HQ177719)和Spodoptera latifascia (HQ177678) 作为外群
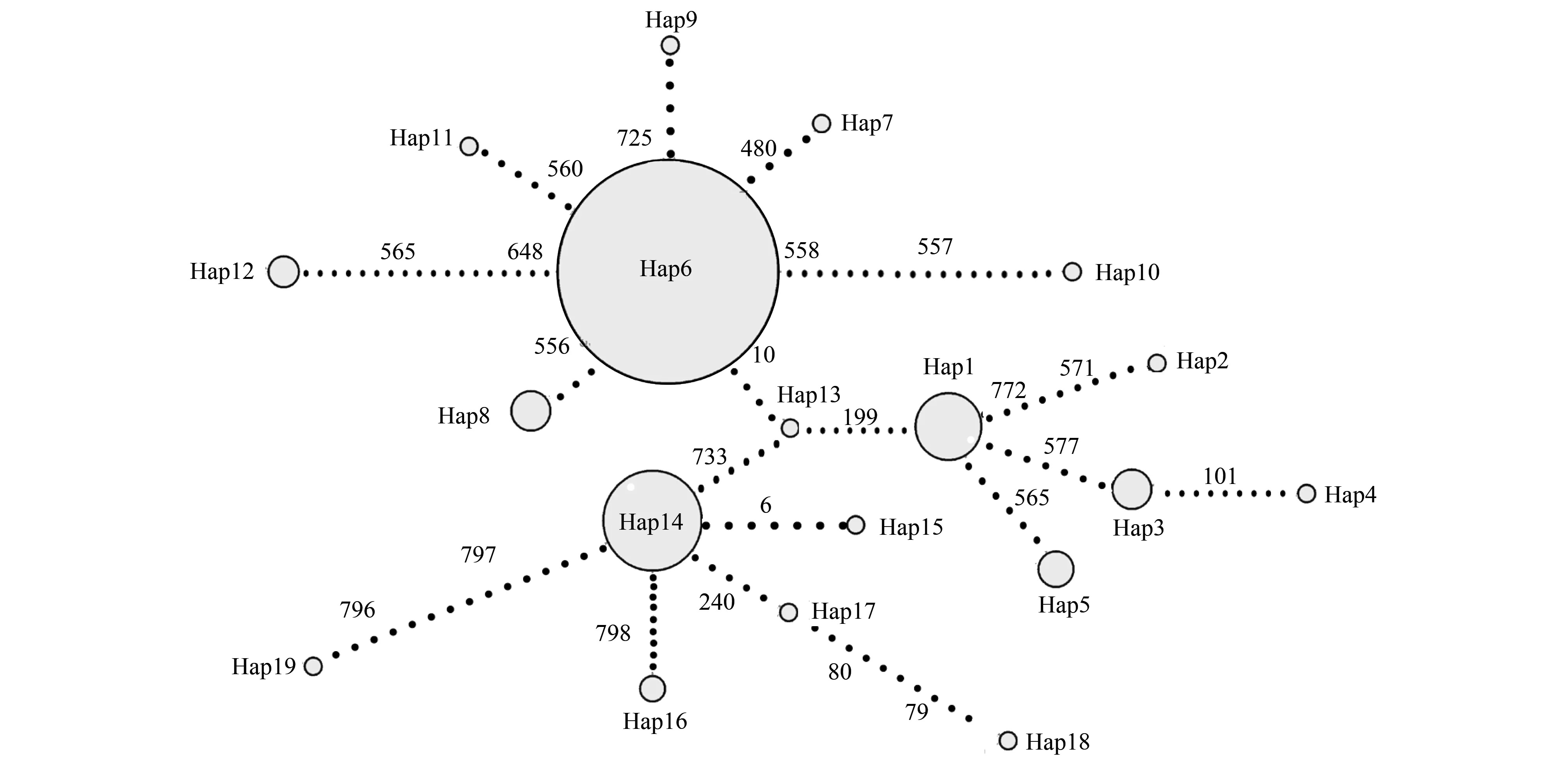
图2 基于mtDNA Cytb基因序列的19个单倍型的Median-joining网络关系图Fig.2 Median-joining network of 19 haplotypes of Spodoptera exigua based on mtDNA Cytb gene sequences每个单倍型用一个圆圈表示,分别代表单倍型Hap1—Hap19;每个圆圈代表一个单倍型,圆的面积与单倍型频率成正比
2.3不同地理种群间遗传距离与遗传分化
研究结果表明,我国北方地区甜菜夜蛾不同地理种群间的平均遗传距离为0.001(0—0.003)。总体上,LUC种群与其它8个地理种群(HLD、SY、HAD、NAY、XINX、YANC、TA和LUC)遗传距离最远,皆为0.003(表3)。另外,19个单倍型之间的遗传距离为0.005(0—0.009)。
遗传分化指数(F-statistic,FST)可表示不同群体间等位基因频率的变异,是反映群体进化历史的重要参数,可在一定程度上揭示种群间基因流和遗传漂变的程度。而基因流则可以揭示出群体间可能的基因渗透及影响遗传分化的遗传现象。Arlequin 3.0计算结果显示,17个地理种群间的遗传分化指数FST为0.108,各地理种群间的遗传分化指数为-0.049—0.666(表3),LUC与其它各地理种群间存在明显的遗传分化(P< 0.05),其余地理种群间的遗传分化程度还较低。
2.4种群遗传结构与种群历史分析
我国北方不同地理种群甜菜夜蛾遗传变异的分子变异分析(AMOVA)结果见表4,不同地理种群间的遗传分化(FST)变异较小(FST= 0.108,P<0.001)。89.18%遗传变异主要发生在种群内,而种群间的变异仅为10.82%,说明我国北方地区甜菜夜蛾的遗传变异主要来自于种群内部,种群间的遗传变异水平较低。
表3基于Kimura 2-parameter遗传距离的种群遗传分化FST值(下三角)和遗传距离(上三角)
Table 3Population pairwiseFSTvalues (below diagonal) and genetic distance (above diagonal) between the populations ofSpodopteraexigua

种群代码PopulationcodeDALFXHLDSYXINLHADNAYXINXYANCBZPENLTAZQLUCYCDLTJDAL0.0010.0010.0010.0010.0010.0010.0010.0020.0020.0010.0010.0010.0020.0010.0010.001FX-0.0030.0010.0010.0010.0010.0010.0010.0020.0020.0010.0010.0010.0020.0010.0010.001HLD0.097*0.069*0.0010.0010.0000.0010.0010.0020.0020.0010.0010.0010.0030.0010.0020.001SY-0.040-0.0410.0500.0010.0000.0010.0010.0020.0020.0020.0010.0010.0030.0010.0020.002XINL0.1110.0260.0330.0390.0000.0000.0000.0010.0010.0010.0010.0010.0020.0010.0010.001HAD0.208*0.1440.0530.122*0.0420.0000.0000.0010.0010.0010.0000.0000.0030.0010.0010.001NAY0.1110.0460.0180.031-0.0300.0000.0000.0010.0020.0010.0000.0010.0030.0010.0010.001XINX0.1110.0260.0330.039-0.0220.031-0.0380.0010.0010.0010.0000.0010.0030.0010.0010.001YANC-0.027-0.0070.120*-0.0350.0370.200*0.117*0.120*0.0020.0020.0020.0010.0030.0020.0020.002BZ0.0230.0010.150*-0.0110.0820.214*0.135*0.134*-0.0110.0020.0020.0020.0020.0020.0020.002PENL-0.0490.0020.086-0.032-0.0040.134*0.082*0.086*-0.0190.0280.0010.0010.0030.0010.0020.002TA0.155*0.119*0.057*0.0860.0130.028-0.0070.0380.183*0.206*0.129*0.0010.0030.0010.0010.001ZQ-0.0260.0050.044-0.022-0.0590.0910.0280.0300.0280.079*-0.0160.0710.0020.0010.0010.001LUC0.376*0.332*0.531*0.286*0.497*0.666*0.558*0.565*0.222*0.153*0.338*0.017*0.0020.0020.003YC-0.059-0.0040.084*-0.039-0.0090.1670.0890.091-0.0380.029-0.0430.134*-0.0290.377*0.0020.001DL0.052-0.010-0.0100.0040.0850.195*0.119*0.105*0.0140.0140.0550.194*0.0870.193*0.0550.002TJ-0.0290.0190.019-0.0330.0420.2790.1470.158-0.0280.017-0.0290.193*0.0240.311*-0.0270.051
*表示差异显著(P<0. 05),Inf代表无穷大
各地理种群进行中性检测结果见表2。检测结果表明,各地理种群无显著差异(P> 0.05),表明大多数地理种群内的序列在进化上遵循中性模型。而将所有个体作为一个整体进行分析,两个中性检验参数(Tajima′sD和Fu′sFS)皆为负值,且达到差异显著水平,说明我国8省甜菜夜蛾作为一个整体种群偏离中性进化,因此,推测我国甜菜夜蛾曾经有种群扩张事件。另外,通过对所有地理种群序列的单倍型错配分布进行分析(图3),表明所有的Cytb单倍型呈现为一条虽不完整平滑但只有一个明显顶峰的单峰型曲线,进一步揭示了甜菜夜蛾曾作为一个整体经历过种群扩张。

表4 不同地理种群甜菜夜蛾遗传变异的分子变异分析(AMOVA)
固定系数 Fixation index,FST= 0.108,***P<0.001

图3 歧点分布与Tajima′s D 和Fu′s FS的中性检验 Fig.3 Mismatch distribution and the results of Tajima′s D and Fu′s FS tests with associated probability for all sampling localities in China实线代表期望值;带有圆环的虚线代表观察值。
3讨论
3.1mtDNACytb基因序列变异与遗传多样性
研究物种遗传变异有助于揭示该物种起源与进化历史[42]。本研究通过分析我国北方不同地理种群甜菜夜蛾Cytb基因片段的遗传变异情况,发现该地区甜菜夜蛾种群呈现出较低遗传多态性,并且,在所研究的Cytb目的片段基因中,仅有22个变异位点。甜菜夜蛾各地理种群间平均遗传距离仅为0.001。与之相一致的是,在其它夜蛾科昆虫中,豹灯蛾Arctiacaja和曲纹灰蝶Lampidesboeticus也得到了相同的结果[34, 43]。较低水平的遗传多样性表明,甜菜夜蛾种群可能经过严重的瓶颈效应或奠基者效应,可能与甜菜夜蛾在我国北方具有较短的种群扩张历史,尚未积累过多的遗传变异有关。而值得注意的是,BZ种群具有相对较高的遗传多样性,可能与该地区甜菜夜蛾没有经过严重的瓶颈效应、奠基者效应或者较小的选择压力(杀虫剂等)有关。
3.2单倍型分布与系统发育分析
基于最大简约树(MP)、最大似然树(ML)与贝叶斯树(BI)构建的单倍型系统发育树,以及基于Median-joining法构建的单倍型网络关系的结果表明,两者结果基本一致,但单倍型网络关系可直接看出单倍型之间的进化关系,尽管MP/ML系统树的后验概率较低,单倍型系统发育树表现出2个分支,但各单倍型中并没有按照地理分布形成明显的族群,各单倍型都相互散布在不同的地理种群中,未形成明显的系统地理结构。LUC、DL、XINX、TA、BZ、YANC、SY和PENL种群所特有的10个独有单倍型,说明各地理种群存在一定基因交流的同时,也具有一定程度的遗传分化。值得注意的是,在所确定的19个单倍型中,单倍型 Hap6是各地理种群中普遍共享的单倍型。通常认为,共享单倍型源于共同的祖先,是一种较为稳定、能够适应环境选择的单倍型[44- 45]。
3.3遗传距离与遗传分化
通常,具有较强扩散能力的物种在不同地区间具有较小的遗传分化。在本研究中,大部分地理种群间没有显著的遗传分化,具有较小序列差异,其结果与牛成伟等一致[27]。这可能归因于群体分化程度主要是对所在生态条件相适应的结果,环境作用强度及方向大体相同,则会造成各分布区内的种群在遗传上将难以形成显著的分化[46]。由于山脉的隔离,例如太行山山脉的隔离可能造成了LUC种群与其它地理种群存在显著的遗传分化。我国8省甜菜夜蛾地理种群间的遗传分化指数FST为0.108,基因流Nm为4.130。当Nm>4时,种群间的基因交流就更为充分,遗传分化更小[47]。可见我国北方甜菜夜蛾各地理种群间基因交流充分,制约了地理种群间的遗传分化。本研究在一定程度上为甜菜夜蛾的系统演化研究提供了分子水平证据,为该种害虫的合理有效控制提供了分子生物学方面的基础资料。
3.4种群遗传结构与种群历史分析
群体遗传结构是指遗传变异在物种或群体中的一种非随机分布,这种非随机分布是由不同过程共同作用产生的,包括物种长期进化历史、分布区改变、生境破碎、群体隔离、突变、遗传漂变、繁育系统、基因流与选择等[43, 46]。因此,深入理解害虫种群遗传结构,有助于提供给我们重要的生物学信息、迁飞规律和地理变异格局[48- 49]。另外。昆虫的扩散能力、地理隔离以及近来人类活动(如苗木运输等)皆可影响物种的种群结构。寄主分布可直接影响专食性昆虫的分布[50]。寄主专化有帮助于保持昆虫种内的高度的种内变异[51- 52],展现出高水平的遗传结构[53- 54]。总体上,除了我国北方甜菜夜蛾没有明显的栖息地丢失、栖息地片段化、寄主不连续分布、气候屏障等影响甜菜夜蛾的存活外,特别是该种害虫的迁飞扩散导致充分的基因流的产生也会使得我国北方不同地理种群的甜菜夜蛾表现出具有较小的遗传分化。
在本研究中,Tajima′sD和Fu′sFS中性进化检测以及歧点分布分析来研究甜菜夜蛾种群历史,显著的两个中性检验参数的负值以及错配分析所呈现的单峰分布都表明甜菜夜蛾近期发生过种群扩张。另外,通常单倍型的星状网络图被认为是种群扩张的证据。在本研究中,单倍型网络图呈星状分布特征,分布格局表明甜菜夜蛾群体过去由于瓶颈效应经历过种群扩张。
3.5害虫防治启示与未来工作
目前,甜菜夜蛾广泛分布于我国各蔬菜主产区。鉴于该种害虫的严重危害性,有必要了解其扩散风险与潜在地理分布。因此,应积极对其进行有效监控,采取一些措施尽量避免其快速扩散到其它蔬菜主产区。抗药性的产生是昆虫适应环境与长期进化的结果,同时也是人为自然选择的结果[52- 53]。在过去的几十年,一些地区大量使用单一杀虫药剂来防治甜菜夜蛾,结果造成了严重的抗药性[13, 54]。同时,也造成一些地区的甜菜夜蛾经历了严重的种群瓶颈效应[55]。大多数敏感性个体被杀死,仅有少量具有抗性的个体存活下来。此外,不同地理种群间充分的基因流可以用来解释抗药性的快速扩散,同时,许多抗性特征也通过长距离的迁移被积累下来[56]。因此,在较大时间和空间尺度调查甜菜夜蛾的抗药性,结合抗性基因与遗传结构分析,将有助于了解甜菜夜蛾的抗性水平与抗药类别,最终指导化学药剂的合理使用。例如,甜菜夜蛾不同地理种群间遗传距离小、分化程度低,说明虫源相似,在进行化学防治时,尽量避免施用与虫源地使用相同的药剂类型,以避免产生的抗药性影响防治效果;对遗传多样性丰富的地区,通常认为甜菜夜蛾具有较高适应环境变化的能力,防治比较困难,需采取轮换施药,降低抗性上升速度。
此外,近年来研究表明,通过母系mtDNA与核基因组中mtDNA假基因间,以及父系渗漏引起的不同单倍型的双亲mtDNA间发生基因重组[57- 58],这对以mtDNA严格母系遗传为基础的许多应用领域产生重要影响[59]。例如,在群体遗传、进化与系统发育研究方面,mtDNA 重组可能影响关于群体大小变化的推论[60]。与此同时,mtDNA重组还会影响系统发育关系构建和分子定时的准确性[61]。因此,为了更好的理解甜菜夜蛾的种群进化历史和种群遗传结构,有必要使用更长、更多的基因序列,甚至是更快的进化标记,如微卫星等[62- 63]。
参考文献(References):
[1]Dingha B N, Moar W J, Apple A G. Effects ofBacillusthuringiensisCry 1C toxin on the metabolic rate of Cry 1C resistant and susceptibleSpodopteraexigua(Lepidoptera: Noctuidae). Physiological Entomology, 2004, 29(5): 409- 418.
[2]Rizwan-ul-Haq M, Hu Q B, Hu M Y, Lin S Q, Zhang W L. Biological impact of harmaline, ricinine and their combined effects withBacillusthuringiensisonSpodopteraexigua(Lepidoptera: Noctuidae). Journal of Pest Science, 2009, 82(4): 327- 334.
[3]Guo J Y, Wu G, Wan F H. Activities of digestive and detoxification enzymes in multiple generations of beet armyworm,Spodopteraexigua(Hübner), in response to transgenic Bt cotton. Journal of Pest Science, 2010, 83(4): 453- 460.
[4]魏娟, 陈浩涛, 崔璟辉, 周生海. 1989—2010年我国甜菜夜蛾文献计量分析. 长江蔬菜: 学术版, 2010, (18): 124- 127.
[5]郑霞林, 王攀, 王小平, 雷朝亮. 大葱甜菜夜蛾主要生物学习性、暴发成因及防治. 长江蔬菜: 学术版, 2009, (18): 4- 7.
[6]朱国仁, 古希树, 王少丽, 张友军, 胡霞, 徐维红. 天津地区大葱甜菜夜蛾发生规律和综合治理. 长江蔬菜: 学术版, 2010, (18): 96- 10.
[7]Adamczyk Jr J J, Williams M R, Reed J T, Hubbard D W, Hardee D D. Spatial and temporal occurrence of beet armyworm (Lepidoptera: Noctuidae) moths in Mississippi. Florida Entomologist, 2003, 86(3): 229- 232.
[8]Feng H Q, Wu K M, Cheng D F, Guo Y Y. Radar observation of the autumn migration of the beet armyworm,Spodopteraexigua, and other moths in northern China. Bulletin of Entomological Research, 2003, 93(2): 115- 124.
[9]Jiang X F, Luo L Z, Sappington T W. Relationship of flight and reproduction in beet armyworm,Spodopteraexigua(Lepidoptera: Noctuidae), a migrant lacking the oogenesis-flight syndrome. Journal of Insect Physiology, 2010, 56(11): 1631- 1637.
[10]Meinke L J, Ware G W. Tolerance of three beet armyworm strains in Arizona to methomyl.Journal of Economic Entomology, 1978, 71(4): 645- 646.
[11]Chaufaux J, Ferron P. Sensibilité différente de deux populations deSpodopteraexiguaHüb. (Lépid.,Noctuidae) aux baculovirus et aux pyréthrinoïdes de synthèse. Agronomie, 1986, 6(1): 99- 104.
[12]Chatterjee S N, Tanushree T. Molecular profiling of silkworm biodiversity in India. Genetika, 2004, 40(12): 1618- 1627.
[13]Behura S K. Molecular marker systems in insects: current trends and future avenues. Molecular Ecology, 2006, 15(11): 3087- 3113.
[14]Avise J C. Ten unorthodox perspectives on evolution prompted by comparative population genetic findings on mitochondrial DNA. Annual Review of Genetics, 1991, 25: 45- 69.
[15]Kim Y, Kim N. Cold hardiness inSpodopteraexigua(Lepidoptera: Noctuidae). Environmental Entomology, 1997, 26(5): 1117- 1123.
[16]Sunnucks P. Efficient genetic markers for population biology. Trends in Ecology& Evolution, 2000, 15(5): 199- 203.
[17]Hurst G D D, Jiggins F M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proceedings of the Royal Society B: Biological Sciences, 2005, 272(1572): 1525- 1534.
[18]Gray M W. Origin and evolution of mitochondrial DNA. Annual Review of Cell Biology, 1989, 5: 25- 50.
[19]Morit A C, Dowling T E, Brown W M. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annual Review of Ecology and Systematics, 1987, 18: 269- 292.
[20]Pirounakis K, Koulianos S, Schmid- Hempel P. Genetic variation among European populations ofBombuspascuorum(Hymenoptera, Apidae) from mitochondrial DNA sequence data. European Journal of Entomology, 1998, 95(1): 27- 33.
[21]Morrow J L, Scott L, Congdon B, Yeates D K,Frommer M, Sved J A. Close genetic similarity between two sympatric species of tephritid fruit fly reproductively isolated by mating time. Evolution: International Journal of Organic Evolution, 2000, 54(3): 899- 910.
[22]Birungi J, Arctander P. Large sequence divergence of mitochondrial DNA genotypes of the control region within populations of the African antelope, kob (Kobuskob). Molecular Ecology, 2000, 9(12): 1997- 2008.
[23]dela Cruz K D, Whiting M F. Genetic and phylogeographic structure of populations ofPulexsimulans(Siphonaptera) in Peru inferred from two genes (CytbandCoII). Parasitology Research, 2003, 91(1): 55- 59.
[24]Ren Z M, Ma N B, Guo Y P. The studies of the phylogeny of acridoidea based on mtDNA sequences. Acta Genetica Sinica, 2002, 29(4): 314- 321.
[25]张艳. 甜菜夜蛾Spodopteraexigua抗性遗传研究及AFLP体系的建立[D]. 长春: 吉林农业大学, 2005.
[26]孙小洁, 李朝飞, 于航, 裘雪梅, 李庆, 张文庆. 甜菜夜蛾cDNA文库的构建. 昆虫知识, 2006, 43(3): 404- 407.
[27]牛成伟, 张青文, 叶志华, 罗礼智. 不同地区甜菜夜蛾种群的遗传多样性分析. 昆虫学报, 2006, 49(5): 867- 873.
[28]Sezonlin M, Dupas S, LeRü B, LeGall P, Moyal P, Calatayud P A, Giffard I, Silvain J F. Phylogeography and population genetics of the maize stalk borerBusseolafusca(Lepidoptera, Noctuidae) in sub-Saharan Africa. Molecular Ecology, 2006, 15(2): 407- 420.
[29]Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P. Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Annals of the Entomological Society of America, 1994, 87(6): 651- 701.
[30]Thompson J D, Gibson T J, Plewniak F, Jeanmougin F, Higgins D G, 1997. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research, 25(24): 4876- 4882.
[31]Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA 5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 2011, 28(10): 2731- 2739.
[32]Kimura M. A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. Journal of Molecular Evolution, 1980, 16(2): 111- 120.
[33]Swofford D L. Paup*: Phylogenetic analysis using parsimony (*and other methods), 4.0 beta 10 ed. Sinauer, Sunderland, Mass, 2002.
[34]Posada D, Crandall K A. MODELTEST: Testing the model of DNA substitution. Bioinformatics, 1998, 14(9): 817- 818.
[35]Posada D, Buckley T R. Model selection and model averaging in phylogenetics: Advantages of akaike information criterion and Bayesian approaches over likelihood ratio tests. Systematic Biology, 2004, 53(5): 793- 808.
[36]Bandelt H J, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 1999, 16(1): 37- 48.
[37]Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 2009, 25(11): 1451- 1452.
[38]Excoffier L, Laval G, Schneider S. Arlequin (version 3. 0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 2005, 1: 47- 50.
[39]Weir B S, Hill W G. EstimatingF- statistics. Annual Review of Genetics, 2002, 36: 721- 750.
[40]Taylor M F J, McKechnie S W, Pierce N, Kreitman M. The lepidopteran mitochondrial control region: structure and evolution. Molecular Biology and Evolution, 1993, 10(6): 1259- 1272.
[41]Cameron S L, Whiting M F. The complete mitochondrial genome of the tobacco hornworm,Manducasexta(Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene, 2008, 408(1/2): 112- 123.
[42]Lohman D J, Peggie D, Pierce N E, Meier R. Phylogeography and genetic diversity of a widespread Old World butterfly,Lampidesboeticus(Lepidoptera: Lycaenidae). BMC Evolutionary Biology, 2008, 8: 301.
[43]Anderson S J, Conrad K F, Gillman M P, Woiwod I P, Freeland J R. Phenotypic changes and reduced genetic diversity have accompanied the rapid decline of the garden tiger moth (Arctiacaja) in the UK. Ecological Entomology, 2008, 33(5): 638- 645.
[44]张立勋, 阮禄章, 安蓓, 刘迺发. 西藏雪鸡青海亚种的种群遗传结构和地理变异. 动物学报, 2005, 51(6): 1044- 1049.
[45]周志军, 张艳霞, 常岩林, 杨明茹. 暗褐蝈螽不同地理种群间的遗传分化. 遗传, 2011, 33(1): 75- 80.
[46]Armstrong K F, Wratten S D. The use of DNA analysis and the polymerase chain reaction in the study of introduced pests in New Zealand // Symondson W O C, Liddell J E, eds. The Ecology of Agricultural Pests. Melbourne: Chapman and Hall, 1996: 231- 263.
[47]Boivin T, Bouvier J C, Beslay D, Suphanor B. Variability in diapause propensity within populations of a temperate insect species: interactions between insecticide resistance genes and photoperiodism. Biological Journal of the Linnean Society, 2004, 83(3): 341- 351.
[48]Miller N J, Birley A J, Overall A D J, Tatchell G M. Population genetic structure of the lettuce root aphid,Pemphigusbursarius(L.), in relation to geographic distance, gene flow and host plant usage. Heredity, 2003, 91(3): 217- 223.
[49]Timm A E, Pringle K L, Warnich L. Genetic diversity of woolly apple aphidEriosomalanigerum(Hemiptera: Aphididae) populations in the Western Cape, South Africa. Bulletin of Entomological Research, 2005, 95(3): 187- 191.
[50]Mendelson T C, Shaw K L. Use of AFLP markers in surveys of arthropod diversity. Methods in Enzymology, 2005, 395: 161- 177.
[51]Avise J C, Arnold J, Ball R M, Bermingham E, Lamb T, Neigel J E, Reeb C A, Saunders N C. Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annual Review of Ecology Evolution and Systematics, 1987, 18: 489- 522.
[52]Karunaratne S H P P. Insecticide resistance in insects: a review. Ceylon Journal of Science (Biological Sciences), 1998, 25: 72- 99.
[53]Ffrench-Constant R H, Daborn P J, Le Goff G. The genetics and genomics of insecticide resistance. Trends in Genetics, 2004, 20(3): 163- 170.
[54]Feyereisen J. Molecular biology of insecticide resistance. Toxicology Letters, 1995, 82- 83: 83- 90.
[55]Saeed Q, Saleem M A, Ahmad M. Toxicity of some commonly used synthetic insecticides againstSpodopteraexigua(Fab) (Lepidoptera: Noctuidae). Pakistan Journal of Zoology, 2012, 44(5): 1197- 1201.
[56]Bouvier J C, Buès R, Boivin T, Boudinhon L, Beslay D, Sauphanor B. Deltamethrin resistance in the codling moth (Lepidoptera: Tortricidae): inheritance and number of genes involved. Heredity, 2001, 87(4): 456- 462.
[57]McVean G A T. What do patterns of genetic variability reveal about mitochondrial recombination? Heredity, 2001, 87(6): 613- 620.
[58]Guo X H, Liu S J, Liu Y. Evidence for recombination of mitochondrial DNA in triploid crucian carp. Genetics, 2006, 172(3): 1745- 1749.
[59]于晓丽, 黄原. 动物线粒体DNA重组的研究进展. 动物学杂志, 2008, 43(2): 145- 149.
[60]Piganeau G, Gardner M, Eyre-Walker A. A broad survey of recombination in animal mitochondria. Molecular Biology and Evolution, 2004, 21(12): 2319- 2325.
[61]Hagelberg E. Recombination or mutation rate heterogeneity? Implications for Mitochondrial Eve. Trends in Genetics, 2003, 19(2): 84- 90.
[62]Posada D, Crandall K A. Intraspecific gene genealogies: trees grafting into networks. Trends in Ecology & Evolution, 2001, 16(1): 37- 45.
[63]Downie D A. Evidence for multiple origins of grape phylloxera (DaktulosphairavitifoliaeFitch) (Hemiptera: Phylloxeridae) in South African vineyards. African Entomology, 2005, 13(2): 359- 365.
Genetic diversity and population history among geographic populations ofSpodopteraexiguain North China based on mtDNACytbgene sequences
WANG Xingya1,*, ZHOU Lihong2
1InstituteofPlantProtection,LiaoningAcademyofAgriculturalSciences,Shenyang110161,China2InstituteofFlowerResearch,LiaoningAcademyofAgriculturalSciences,Shenyang110161,China
Abstract:The beet armyworm, Spodoptera exigua (Lepidoptera: Noctuidae), is an important and cosmopolitan pest that attacks cultivated crops, including maize, cotton, soybeans, beet, tomato, cabbage, and alfalfa, causing serious economic losses in some of the main crop-producing areas. This species originated in South Asia and is distributed throughout the tropical and temperate regions of Asia, Europe, Africa, and North America. In China, S. exigua was first recorded in Beijing in the 1890s and has been widely distributed in the southern provinces in recent years. However, recently, with global climate warming and the adjustment of agricultural planting structures, S. exigua has quickly spread to the main crop-producing areas of North China. Genetic diversity and population genetic structure are important aspects of the population genetics of agricultural insects, and provide essential information for understanding local adaptation and dispersal patterns, and for clarifying the relationships between genetic variation and outbreaks of pest species. Cytochrome b (Cytb) has a moderate evolutionary rate and a clear evolutionary pattern, suitable for the studies of phylogenetic evolution at the intra- and inter-specific levels. To identify the genetic diversity and population history among geographic populations of S. exigua in North China, and clarify its population demographic history and genetic structure, the genetic diversity, structure, differentiation, and molecular variance were analyzed using DnaSP 5.0 and Arlequin 3.0. In the present study, 798 bases of mtDNA Cytb genes were obtained from 304 individuals of S. exigua, collected from 17 geographic populations in 8 provinces of North China in 2012. Of the 798 characters, 776 sites were conserved and 22 sites were variable (2.8% of the total length), including 9 parsimonious informative sites. The average base frequencies were 42.7% A, 33.1% T, 12.6% G, and 11.5% C. Within-locality diversity was estimated in terms of haplotype diversity (h) and nucleotide diversity (π) from all surveyed localities. Low genetic diversity (Hd = 0.422 ± 0.035,π = 0.00119 ± 0.00011) in the total populations among different geographic populations was detected. The highest estimate (Hd = 0.643 ± 0.106, π = 0.00214 ± 0.00039) was detected in Binzhou (BZ). Nineteen haplotypes, including 9-shared haplotypes were identified. Hap6 was shared in 228 individuals of all populations. Phylogenetic analysis was conducted to determine the relationships between S. exigua haplotypes, and detect discernible groups related to geographic distribution. High congruence was observed between the phylogenies derived from Maximum parsimony (MP), Maximum-likelihood (ML) and Bayesian analyses, and these analyses generated only two inclusive clades. Moreover, the median-joining network was similar to the topology of the phylogenetic tree with 19 haplotypes, and revealed no obvious phylogeographic pattern. The pairwise FSTvalues between the populations varied from -0.049—0.666. Generally, little genetic differentiation (FST= 0.108, P<0.001) among different geographic populations was detected, with the only significant differentiation between the Lucheng (LUC) and other S. exigua populations. The analysis of molecular variance (AMOVA) showed that the percentage of variation within a population (89.18%) was greater than that between the populations (10.82%). The results of neutrality tests on the S. exigua data set of the total population are: Tajima′s D (D = -1.897, P<0.05) and Fu′s Fs (Fs = -4.424, P<0.05), combined with the unimodal mismatch distribution, indicated recent population expansion of S. exigua in large spatial scales.
Key Words:Spodoptera exigua; cytochrome b (Cytb); mtDNA; population history; genetic differentiation
基金项目:国家自然科学基金项目(31101626)
收稿日期:2014- 10- 04; 网络出版日期:2015- 08- 21
*通讯作者
Corresponding author.E-mail: wangxingya20081@163.com
DOI:10.5846/stxb201410041952
王兴亚,周俐宏.基于mtDNACytb基因序列的我国北方地区甜菜夜蛾遗传多样性与种群历史分析.生态学报,2016,36(8):2337- 2347.
Wang X Y, Zhou L H.Genetic diversity and population history among geographic populations ofSpodopteraexiguain North China based on mtDNACytbgene sequences.Acta Ecologica Sinica,2016,36(8):2337- 2347.
