
微滴数字PCR定量检测全血样品中结核分枝杆菌特异CFP10基因
2016-07-12 17:21:06宋能谭杨罗凤玲关清闫明哲潘勤章晓联
中国生化药物杂志 2016年2期
宋能,谭杨,罗凤玲,关清,闫明哲,潘勤,章晓联Δ
(1.武汉大学 病毒学国家重点实验室/湖北省过敏及免疫相关疾病重点实验室/武汉大学医学研究院/武汉大学基础医学院 免疫学系,湖北 武汉 430071;2.武汉市医疗救治中心,湖北 武汉 430071)
微滴数字PCR定量检测全血样品中结核分枝杆菌特异CFP10基因
宋能1,谭杨1,罗凤玲1,关清1,闫明哲2,潘勤1,章晓联1Δ
(1.武汉大学 病毒学国家重点实验室/湖北省过敏及免疫相关疾病重点实验室/武汉大学医学研究院/武汉大学基础医学院 免疫学系,湖北 武汉 430071;2.武汉市医疗救治中心,湖北 武汉 430071)
目的 采用微滴数字PCR技术(droplet digital PCR, ddPCR)检测全血中结核分枝杆菌特异性CFP10(10-kDa culture filtrate protein,CFP10)基因的拷贝数含量。方法 收集10例活动性肺结核患者(active tuberculosis,aTB)、10例肺外结核患者(extrapulmonary tuberculosis,EPTB)和5例健康对照者(healthy donars , HDs)全血并提取DNA,优化引物最佳扩增条件并制定重组质粒标准曲线,对样本中CFP10基因的拷贝数含量分别用ddPCR与实时定量PCR(quantitative real-time PCR,qPCR)进行检测和比较。 结果 对重组质粒的灵敏度检测显示ddPCR技术明显优于qPCR技术。对样本的检测显示qPCR技术检测aTB、EPTB患者与健康人的全血中CFP10含量之间无统计学意义;但是ddPCR技术检测aTB、EPTB患者与健康人之间有显著统计学意义(P<0.0001); ddPCR检测aTB和EPTB患者(共20例)CFP10 基因绝对定量的浓度约为3.4~94.0 copies/μL。结论 本研究首次报道采用ddPCR技术能灵敏地用于TB患者全血的诊断。
微滴数字PCR; 结核分枝杆菌;定量检测;CFP10
结核分枝杆菌(Mycobacteriumtuberculosis,MTB),俗称结核杆菌,是引起人结核病的主要病原菌。结核病目前仍是很多发展中国家需要面临的公共健康问题。据世界卫生组织(WHO)2012年调查报告显示,全球每年约有130万人死于结核病[1]。目前全球有将近1/3的人感染过结核分枝杆菌(Mycobacteriumtuberculosis,MTB),感染人数以每年800~1000万的速度增加,每年有近300万人死于结核病[2]。我国活动性肺结核人数高居世界第二位,是结核病高负担国家[1]。由于抗结核药物的广泛使用,多重耐药结核杆菌 (multidrug-resistant tuberculosis,MDR-TB)种类不断增多,与此同时艾滋病与结核病联合发病使得结核病的治疗难度加大[3-6]。因而早期而明确的诊断对于结核病的治疗、预后及传播的控制具有极其重要的意义[7]。
目前临床上应用的结核病诊断技术主要有结核菌素纯蛋白衍生物(protein purified derivative,PPD)试验、痰培养、抗酸染色、血清学抗体检测方法[8], 但是各有灵敏度低、耗时、或特异性差等缺点;采用痰涂片等抗酸染色检测的检出率仅约为33%[9]。由于血液中结核分枝杆菌含量极低,其难以血液样本中结核基因进行准确检测, 一般技术很难检测到[10]。最近发展的实时定量PCR(quantitative real-time PCR,qPCR)技术、干扰素(interferon-γ,IFN-γ)的酶联免疫斑点技术(enzyme-linked immunospot, ELISPOT)具有较快速、灵敏度高、特异性较强的优点, IFN-γ的ELISPOT用于检测结核患者外周血中淋巴细胞经结核特异抗原CFP10和早期分泌性抗原(6-kDa early secreted antigenic target,ESAT-6) 刺激后释放的IFN-γ水平; 但是qPCR技术检测的是结核特异基因的含量,样本通常仅限于患者痰液而不是血液(由于血液中结核菌含量很低), 或对少数免疫低下患者血液(血液中结核菌含量高于正常人含量)的检测[11], 由于痰液取样均一性较差等因素造成诊断准确性下降。 qPCR要求目的基因拷贝数相对较高,对目的基因做出相对定量分析[10], 会出现假阳性(如探针保存不当而部分降解)和假阴性结果(对于低拷贝数时)。并且在分析外源基因拷贝数时必须依赖于标准曲线和已知拷贝数的基因,而标准曲线的制备容易受各种因素的影响,故此法对诊断标准的统一性仍值得商榷[12]。
微滴数字PCR(droplet digital PCR, ddPCR)是近年来兴起的一种新的绝对定量的更灵敏技术,又称为第三代PCR技术,是通过将微量样品作大倍数稀释和分液(partitioning) , 通过极度稀释实现理论上的单分子扩增,然后用终点法PCR 和统计学中的泊松分布原理计算出样品的原始浓度[13]。与qPCR方法相比,ddPCR兼具qPCR方法的优点,其相对qPCR具有更高的灵敏度和更多元分析的能力,为临床诊断提供独特的分析优势,并且也不需要标准曲线的制备[14-16]。ddPCR已经报道用于检测拷贝数很低的血液中HBV(hepatitis B virus,HBV)的检测[17],在复杂的异质样品中的稀有序列检测方面具有明显的技术优势,能够完成很多qPCR无法完成的检测任务。目前,ddPCR已被用于病毒的分析及肿瘤分子标志物稀有突变的检测,与疾病相关的基因拷贝数变异检测以及其他病原微生物的RNA检测和基因表达分析等[18-20]。但是用于结核的诊断还未见报道,因而本研究采用ddPCR对结核患者全血DNA中结核特异CFP10基因拷贝数进行检测。
本研究以aTB患者、EPTB患者和健康人全血DNA为研究对象,选取结核分枝杆菌保守基因Rv3874(亦称CFP10)基因序列,设计相应引物及荧光探针,利用ddPCR方法定量检测目的基因的含量,并与qPCR法进行平行比较; 并采用 ddPCR法对结核患者全血DNA中CFP10拷贝数含量进行绝对定量的检测,以期探讨用于结核病的诊断。
1 材料与方法
1.1 材料
1.1.1 全血及患者来源:10例活动性肺结核患者(active tuberculosis, aTB)与10例肺外结核患者(extrapulmonary tuberculosis, EPTB)(HIV感染者已排除)的EDTA-Na2抗凝全血于2015 年7月在湖北省武汉市医疗救助中心检验科收集。5名健康自愿者来自于武汉大学中南医院。
1.1.2 菌株与试剂:引物及探针由(Invitrogen公司,美国)合成;pET-28a(+)-CFP10由本实验室构建并保存);结核分枝杆菌标准株H37Rv由武汉大学ABSL-Ⅲ实验室保存并提供;大肠埃希菌E.coliDH5α·感受态菌种、pET-28a(+) 质粒由本实验室保存;限制性内切酶EcoRI、BamHI(Fermentas公司,美国);Ligation high高效连接酶(Toyobo公司,日本);全血DNA提取试剂盒、DNA 凝胶回收试剂盒、质粒提取试剂盒、细菌基因组提取试剂盒(Axygen公司,美国);DNA Marker购于东盛生物公司。其他材料:TaqMan® Gene Expression Master Mix (ABI公司, 美国)、ddPCRTMSupermix for Probes、Droplet Generation Oil、Droplet Reader Oil (Bio-Rad公司, 美国)。
1.1.3 主要仪器:DU-730紫外分光光度计(Beckman公司,美国);GelDoc-It310紫外凝胶成像系统(UVP Bioimaging system公司,美国);PowerPacBasic DNA凝胶电泳仪(Bio-Rad公司 ,美国);Pro-s PCR仪(Eppendorf公司, 德国);Step One Plus 实时荧光定量PCR仪(ABI,美国);Bio-Rad S1000 PCR仪、微滴数字PCR仪为QX 100TMDroplet Digital PCR 系统( Bio-Rad公司,美国,包括微滴生成仪、微滴读取仪和QuantaSoft1.7.4结果分析软件)。
1.2 方法
1.2.1 引物和探针的设计及优化:根据结核分枝杆菌CFP10基因序列设计引物及探针,其序列分别为:上游引物P1:5’-AAGCAGCCAATAAGCAGAAGC-3’;下游引物P2:5’-AGCCCATTTGCGAGGACA-3’;探针:5’-FAM-GACGAATATTCGTCAGGCCGG-BQ1-3’。采用设计好的引物和探针建立qPCR方法,包括扩增循环条件的优化及引物和探针浓度的优化。上游引物与下游引物在PCR反应体系中的浓度分别为350 nmol/L,设置探针浓度分别为100、150、200、250、300、350、400和450 nmol/L,加入10 μL 2×TaqMan®Gene Expression Master Mix,pET-28a(+)-CFP10模板2 μL,用超纯水补足至20 μL,然后进行qPCR扩增。扩增条件确定为: 95 ℃,5min预变性;95 ℃,15 s; 60 ℃,30 s,40个循环。
1.2.2 绘制标准曲线:qPCR反应体系和条件:qPCR反应体系为20 μL,包含10 μL 2×TaqMan®Gene Expression Master Mix,10 nmol/L 正向引物和反向引物各1 μL,10 nmol/L探针0.5 μL,DNA模板1 μL,用超纯水补足至20 μL。qPCR使用2步扩增法,程序如下:94 ℃,5 min预变性;94 ℃变性15 s,60 ℃退火60 s,共40个循环。每个模板重复3个平行扩增,重复3 轮实验。扩增结束后使用Step One Plus自带软件进行分析实验数据,并获得qPCR标准曲线,R2值以及扩增效率。ddPCR反应体系和条件:ddPCR反应包括4个步骤:配制体系、生成微滴、扩增循环和信号读取[21]。配制体系使用10 μL 2×ddPCRTMSupermix for Probes,正、反向引物各1 μL ,探针0.5 μL ,DNA模板1 μL,用超纯水补足至20 μL。将配制好的20 μL PCR反应液,转移至微滴发生卡(DG8 cartridge)孔中,再加入70 μL微滴发生油(droplet generation oil)至oil孔中,利用QX100TM微滴式数字PCR仪的微滴生成器制备反应微滴。将每个样品的微滴分别转移至96孔PCR反应板中对应的反应孔中,用铝膜热封(180 ℃,5 sec)后,于普通PCR仪上进行扩增,本实验的PCR扩增是在Bio-Rad S1000 PCR仪上完成的,其中退火温度是经过温度梯度优化后的温度。
ddPCR扩增使用3步法,设置程序如下:95 ℃,10 min预变性;94 ℃变性30 s,60 ℃退火60 s,共45个循环,扩增结束后进行98 ℃、10 min的热变性。每个模板重复3个平行检测。将PCR扩增后的96孔板放入QX100TM微滴式数字PCR仪的微滴分析器中,检测FAM的荧光信号,96个样品检测用时约3 h。仪器会自动分析样品的各微滴荧光信号,然后由QuantaSoft1.7.4完成对数据的自动处理。将重组质粒pET-28a(+)-CFP10进行梯度倍比稀释:109、108、107、106、105、104、103、102和10 copies/μL,各取以上倍比稀释的重组质粒DNA 2 μL分别用于qPCR和ddPCR,并将结果绘制出标准曲线。
1.2.3 qPCR法对样本的检测 应用全血DNA提取试剂盒提取待检测者的全血DNA,经由紫外分光光度计测量OD260/OD280和琼脂糖凝胶进行质量鉴定,DNA浓度为20 ng/μL,OD260/OD280=1.82。全血DNA的提取按Axygen公司试剂盒说明书操作。qPCR反应体系为20 μL,包含10 μL 2×TaqMan® Gene Expression Master Mix,10 nmol/L 正向引物和反向引物各1 μL,10 nmol/L探针0.5 μL ,DNA样本2 μL,用超纯水补足至20 μL。qPCR 使用2步扩增法(同2.2中qPCR 2步扩增法)。扩增结束后使用Step One Plus自带软件获得实验数据。数据采用SPSS17.0统计分析软件进行分析。
1.2.4 ddPCR法对样本的检测 配制体系使用10 μL 2×ddPCRTMSupermix for Probes,正、反向引物各1 μL ,探针0.5 μL,DNA样本2 μL(此样本与方法2.3中所用样本相同),用超纯水补足至20 μL。ddPCR 扩增使用3步法(同2.2中ddPCR 3步扩增法)。将PCR扩增后的96孔板放入QX100TM微滴式数字PCR仪的微滴分析器中,检测FAM的荧光信号,然后由Quanta Soft1.7.4完成对数据的自动处理。

2 结果
2.1 PCR探针最佳反应浓度的确定 通过qPCR扩增得到当探针浓度为350 nmol /L时,荧光信号强度较高,且不再随着浓度的增加而增加 ,见图1。因而后续的PCR反应采用的探针浓度均是350 nmol /L。

图1 qPCR与ddPCR探针最佳浓度的确定 Fig.1 Determination of optimum concentration of probe for qPCR and ddPCR
2.2 qPCR探针法与ddPCR法对CFP10重组质粒DNA的线性范围分析 将pET-28a(+)-CFP10 重组质粒DNA的浓度进行梯度倍比稀释:109、108、107、106、105、104、103、102、10和1copies/μL分别对应S’10-S’1,结果显示当重组质粒浓度达到102copies/μL以下时已偏离标准曲线,说明qPCR最低检测浓度是102copies/μL。 当质粒浓度在108、107、106、105、104、103、102copies/μL时qPCR对目的基因的检测能准确反映出相应浓度标准曲线为:y=-3.324x+1.6459,R2=0.9997,扩增效率(Efficiency)=99.91%,该qPCR标准曲线对重组质粒测定浓度线性范围为:109~102copies/μL,Ct值测定范围为5.049~30.124,见图2。

图2 qPCR法对pET-28a(+)-CFP10重组质粒线性范围的测定Fig.2 Linearity range of qPCR for quantifying pET-28a(+)-CFP10 genomic DNA
ddPCR的标准曲线: 将重组质粒pET-28a(+)-CFP10进行梯度倍比稀释: 107、106、105、104、103、102、10和1.2 copies/μL对应图中S8~S1,各取以上倍比稀释的重组质粒DNA 2 μL用于ddPCR定量分析,其相关方程式为:y=0.9706x+0.0949,R2=0.9987,见图3。结果显示当CFP10重组质粒浓度稀释达到1.2 copies/μL时仍保持着正相关性,说明ddPCR最低检测浓度是1.2 copies/μL以下,因而其灵敏度高于qPCR的最低检测浓度(102copies/μL)。

图3 ddPCR法对pET-28a(+)-CFP10重组质粒线性范围的测定Fig.2B Linearity range of ddPCR for quantifying pET-28a(+)-CFP10 genomic DNA
2.3 qPCR对样本的分析 图4显示10例aTB患者样本 (图4A)与10例EPTB患者(图4B)目的基因CFP10的CT值分布为30.163~32.478均不在制定的标准曲线范围内,故无法通过标准曲线来准确计算出其拷贝数,因而采用相对定量计算出10例aTB病人相对拷贝数为(2.948±0.5375)copies/μL,10例EPTB病人相对拷贝数(9.752±3.755)copies/μL,并分别与5例健康人(healthy donars,HDs)(1.455±0.1526)copies/μL比较算得t值分别为(t=1.929、t=1.549),差异无统计学意义。用相对定量估算出各例病人其相对拷贝数值为1.47~8.8 copies/μL,见图4。
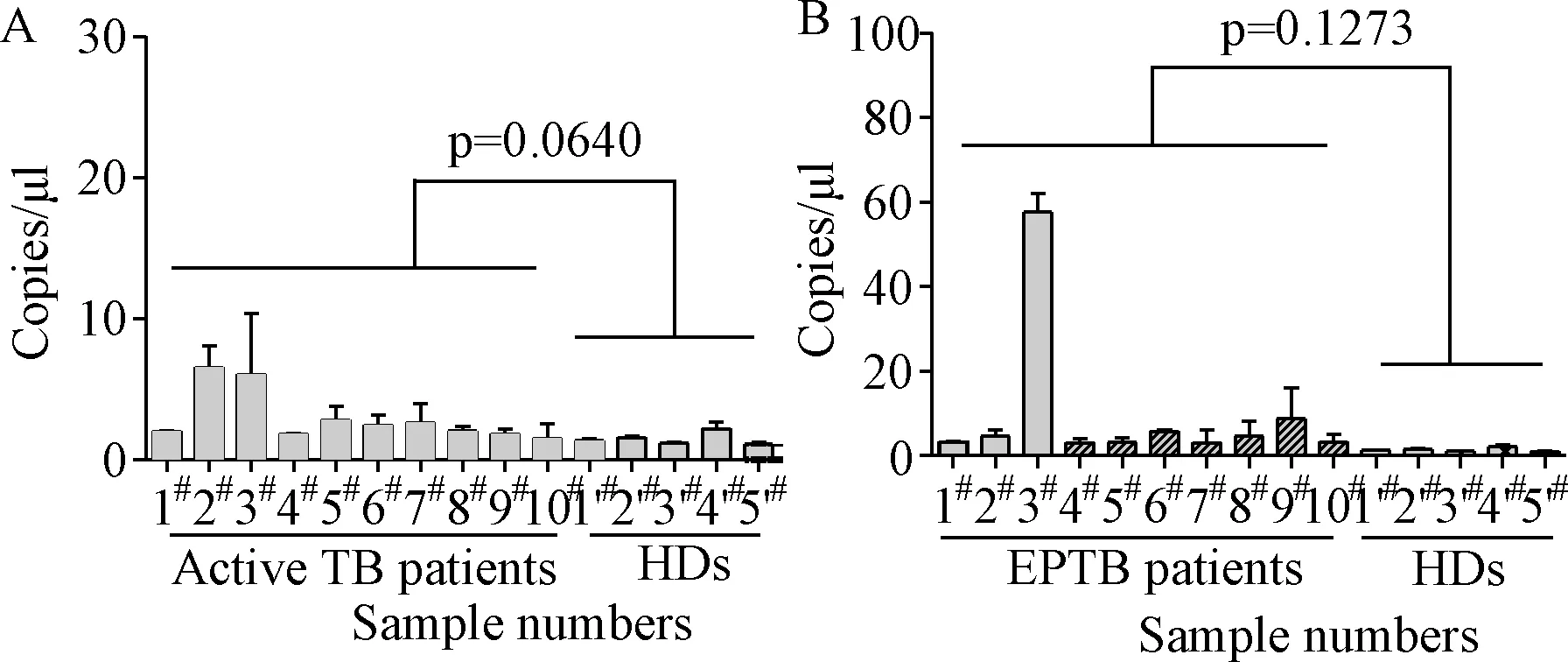
图4 qPCR法对10例aTB病人(A)10例EPTB病人(B)全血DNA中CFP10基因拷贝数的检测Fig.4 qPCR analysis for CFP10 gene copies of whole blood DNA of aTB patients(A)and EPTB patients(B)
2.4 ddPCR对样本的分析 在图5A中(a)显示3号EPTB患者的原始散点图,其阳性微滴数值Pos: 1236, 说明是强阳性;(b)显示了8号aTB患者的原始散点图,其阳性微滴数值Pos: 36, 说明是弱阳性; (c)显示的是健康人阴性对照散点图; (d)显示无模板阴性对照散点图;(e)显示阳性对照(CFP10重组质粒)散点图;(f)显示所有样本综合散点图。图5B、C结果表明10例aTB病人拷贝数(16.47±1.942)copies/μL,10例EPTB病人相对拷贝数(44.99±5.586 )copies/μL,并分别与5例健康人(HDs)对照(0.1740±0.0267)copies/μL比较,算得t值分别为t=5.881与t=5.623,差异有显著统计学意义(P<0.0001)。ddPCR检测aTB和EPTB病人(共20例)CFP10基因绝对定量的浓度范围约为3.4~94.0 copies/μL,见图5。

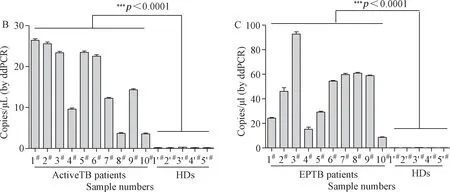
图5 ddPCR法对TB患者样本全血DNA中CFP10基因拷贝数的检测A.原始微滴图片 a :强阳性病例(94.0 copies/μL) ; b:弱阳性病例(3.4 copies/μL) ; c:临床阴性样本; d:阴性对照; e:阳性对照; f:各样本及对照综合B. ddPCR法对10例aTB患者全血DNA中CFP10基因拷贝数检测;C.ddPCR法对10例EPTB患者全血DNA中CFP10基因拷贝数检测Fig.5 Measurement of CFP10 gene copies from whole blood samples of TB patients by ddPCR analysisA. Part of the original micro drop pictures, a: The strong positive cases copies/mu (94.0 copies/μL); b:Weak positive cases copies/mu (3.4 copies/μL); c: Clinical negative samples; d: Negative control; e:Positive control; f:All samples and comprehensive;B. ddPCR analysis for CFP 10 gene copies from whole blood DNA of aTB patients; C. ddPCR analysis for CFP10 gene copies from whole blood DNA of EPTB patients
3 讨论
目前临床上应用的结核病诊断技术多种多样, 当靶标序列含量很低时采用qPCR检测技术往往无法获得准确的结果,因此需要更加灵敏的方法检测低含量的DNA。ddPCR作为一种新兴的、准确的定量技术,正大力推广研究于DNA的绝对定量检测中,尤其是对微量样本的绝对定量[23-24]。目前国内外有见报道通过ddPCR技术对病原微生物的检测有很多,例如用于检测拷贝数很低的血液中HBV的检测[17]、对HIV感染者的检测[25]、对沙眼衣原体的检测[26],植物病原菌的检测[27]、手足口病的检测[28]等,在对AIDS患者血液中HIV病毒检测报道的核酸拷贝数可低至2 copies/μL,对HBV患者石蜡包埋肝组织提取的总DNA中能检测到HBV病毒核酸拷贝数能低至6 copies/μL[17],但对结核杆菌基因组的检测还未见报道。本研究报道通过标准曲线采用ddPCR能检测到CFP10拷贝数能低至1.2 copies/μL(图3)。
本研究选取了编码结核分枝杆菌CFP10基因属于其RD1 (Region of deletion 1)区的DNA序列,RD1区基因存在于结核分枝杆菌标准株(M.tuberculosisH37Rv)、非洲分枝杆菌(M.africanum)、牛分枝杆菌(M.bovis)等有毒菌株中。耻垢分枝杆菌(M.smegmatis)、H37Ra中无此区域。基于ESAT-6、CFP10蛋白的IFN-γ的T-SPOT是世界范围内公认的结核病确诊的最重要诊断手段,在临床检测方面有独特的优势,故选取检测CFP10基因,其特异度强。ddPCR法对结果的判读是基于反应终点阅读到的微滴的荧光信号强度,结合无模板对照和阴阳性对照划出相应的阀值,信号强度位于阀值之上的微滴判定为阳性微滴,位于阀值之下的微滴判定为阴性微滴,并利用泊松分布统计学公式计算出目的DNA分子浓度。本实验选取10例aTB患者和10例EPTB患者全血DNA用于ddPCR检测与分析TB患者全血DNA测得特异性序列浓度范围约为3.4 copies/μL ~ 94.0 copies/μL,以上结果表明,采用qPCR技术检测aTB、EPTB患者与健康人的全血中结核特异基因CFP10含量之间无统计学意义;但是ddPCR技术检测aTB、EPTB患者与健康人之间有显著统计学意义(P<0.0001)。
ddPCR微滴化的过程可以实现靶标序列的富集,从而实现微量靶标序列的检测,即使在低丰度靶标序列检测时,也表现出较好的重复性。ddPCR技术与目前临床上其他结核诊断技术相比具有快速、简便、灵敏和特异性高的优点,并能准确监测TB患者全血中结核杆菌特异性基因的绝对含量。综上所述,ddPCR在结核病的全血诊断中具有很大应用前景。
[1] Glaziou P, Sismanidis C, Floyd K, et al. Global epidemiology of tuberculosis[J]. Cold Spring Harb Perspect Med,2015,5(2):a17798.
[2] Lienhardt C, Glaziou P, Uplekar M, et al. Global tuberculosis control: lessons learnt and future prospects[J]. Nat Rev Microbiol,2012,10(6):407-416.
[3] Li WG, Zhao L, Zhao H. Epidemiology of HIV-Associated Tuberculosis in Urumqi, China[J]. Transplant Proc,2015,47(8):2456-2459.
[4] Kingkaew N, Sangtong B, Amnuaiphon W, et al. HIV-associated extrapulmonary tuberculosis in Thailand: epidemiology and risk factors for death[J]. Int J Infect Dis,2009,13(6):722-729.
[5] Glaziou P, Floyd K, Raviglione M. Global burden and epidemiology of tuberculosis[J]. Clin Chest Med,2009,30(4):621-636.
[6] Gao J, Zheng P, Fu H. Prevalence of TB/HIV co-infection in countries except China: a systematic review and meta-analysis[J]. PLoS One,2013,8(5):e64915.
[7] Vittor AY, Garland JM, Schlossberg D. Improving the diagnosis of tuberculosis: From QuantiFERON to new techniques to diagnose tuberculosis infections[J]. Curr HIV/AIDS Rep,2011,8(3):153-163.
[8] Mitchison DA. The diagnosis and therapy of tuberculosis during the past 100 years[J]. Am J Respir Crit Care Med,2005,171(7):699-706.
[9] 全国结核病流行病学抽样调查技术指导组. 2000年全国结核病流行病学抽样调查报告[J]. 中国防痨杂志,2002(2):3-46.
[10] Lima JF, Guedes GM, Lima JF, et al. Single-tube nested PCR assay with in-house DNA extraction for Mycobacterium tuberculosis detection in blood and urine[J]. Rev Soc Bras Med Trop,2015,48(6):731-738.
[11] Getahun H, Gunneberg C, Granich R, et al. HIV infection-associated tuberculosis: the epidemiology and the response[J]. Clin Infect Dis,2010,50( Suppl 3):S201-S207.
[12] Cankar K, Stebih D, Dreo T, et al. Critical points of DNA quantification by real-time PCR--effects of DNA extraction method and sample matrix on quantification of genetically modified organisms[J]. BMC Biotechnol,2006(6):37.
[13] Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number[J]. Anal Chem,2011,83(22):8604-8610.
[14] Sedlak RH, Cook L, Huang ML, et al. Identification of chromosomally integrated human herpesvirus 6 by droplet digital PCR[J]. Clin Chem,2014,60(5):765-772.
[15] Zhang BO, Xu CW, Shao Y, et al. Comparison of droplet digital PCR and conventional quantitative PCR for measuring gene mutation[J]. Exp Ther Med,2015,9(4):1383-1388.
[16] Yang R, Paparini A, Monis P, et al. Comparison of next-generation droplet digital PCR (ddPCR) with quantitative PCR (qPCR) for enumeration of Cryptosporidium oocysts in faecal samples[J]. Int J Parasitol,2014,44(14):1105-1113.
[17] Huang JT, Liu YJ, Wang J, et al. Next generation digital PCR measurement of hepatitis B virus copy number in formalin-fixed paraffin-embedded hepatocellular carcinoma tissue[J]. Clin Chem,2015,61(1):290-296.
[18] Jones M, Williams J, Gartner K, et al. Low copy target detection by Droplet Digital PCR through application of a novel open access bioinformatic pipeline, 'definetherain'[J]. J Virol Methods,2014(202):46-53.
[19] Zhao H, Wilkins K, Damon IK, et al. Specific qPCR assays for the detection of orf virus, pseudocowpox virus and bovine papular stomatitis virus[J]. J Virol Methods,2013,194(1-2):229-234.
[20] Brunetto GS, Massoud R, Leibovitch EC, et al. Digital droplet PCR (ddPCR) for the precise quantification of human T-lymphotropic virus 1 proviral loads in peripheral blood and cerebrospinal fluid of HAM/TSP patients and identification of viral mutations[J]. J Neurovirol,2014,20(4):341-351.
[21] Pinheiro LB, Coleman VA, Hindson CM, et al. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification[J]. Anal Chem,2012,84(2):1003-1011.
[22] Morisset D, Stebih D, Milavec M, et al. Quantitative analysis of food and feed samples with droplet digital PCR[J]. PLoS One,2013,8(5):e62583.
[23] Sedlak RH, Jerome KR. Viral diagnostics in the era of digital polymerase chain reaction[J]. Diagn Microbiol Infect Dis,2013,75(1):1-4.
[24] Mazaika E, Homsy J. Digital Droplet PCR: CNV Analysis and Other Applications[J]. Curr Protoc Hum Genet,2014(82):7-24.
[25] Persaud D, Gay H, Ziemniak C, et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant[J]. N Engl J Med,2013,369(19):1828-1835.
[26] Roberts CH, Last A, Molina-Gonzalez S, et al. Development and evaluation of a next-generation digital PCR diagnostic assay for ocular Chlamydia trachomatis infections[J]. J Clin Microbiol,2013,51(7):2195-2203.
[27] Dreo T, Pirc M, Ramsak Z, et al. Optimising droplet digital PCR analysis approaches for detection and quantification of bacteria: a case study of fire blight and potato brown rot[J]. Anal Bioanal Chem,2014,406(26):6513-6528.
[28] Lui YL, Tan EL. Droplet digital PCR as a useful tool for the quantitative detection of Enterovirus 71[J]. J Virol Methods,2014(207):200-203.
(编校:王冬梅)
Sensitive and quantitative detection of specific CFP10 gene ofMycobacteriumtuberculosisin whole blood samples by droplet digital PCR
SONG Neng1, TAN Yang1, LUO Feng-ling1, GUAN Qing1, YAN Ming-zhe2, PAN Qin1, ZHANG Xiao-lian1Δ
(1.State Key Laboratory of Virology/Hubei Province Key Laboratory of Allergy and Immunology/ Medical Research Institute,Wuhan University/Department of Immunology, Wuhan University School of Basic Medical Sciences, Wuhan 430071, China; 2.Wuhan Medical Treatment Center, Wuhan 430071, China)
ObjectiveTo analyse content of mycobacterium tuberculosis (MTB) specific CFP10 gene copies from whole blood samples using droplet digital PCR (ddPCR) technique.MethodsWhole blood samples were collected from 10 activetuberculosis(aTB) patients, 10 extrapulmonarytuberculosis (EPTB)patients and 5 healthy donors (HDs), respectively. Total DNA from each blood samples were extracted. The optimal amplification conditions of the primers were optimized and the standard curves of CFP10 recombinant plasmids were established. 10-kDa culture filtrate protein (CFP10) gene copies from whole blood samples were determined and compared using ddPCR and quantitative real-time PCR (qPCR) techniques.ResultsResults showed that ddPCR had much higher sensitivity for measuring CFP gene than qPCR technique.Compared withHDs, there were significantly statistical differences between aTB and EPTB patients using ddPCR(P<0.0001), however there was no statistical significance using qPCR(P>0.05); The absolute quantitative concentration ranges of CFP10 gene in these 20 TB patients measured by ddPCR were about 3.4~94.0copies/μL.ConclusionsThis is the first report showing that ddPCR technique could used for diagnosis of TB patients from whole blood samples with high sensitive.
droplet digital PCR;Mycobacteriumtuberculosis(MTB); Tuberculosis (TB); CFP10
国家重大传染病专项(2012ZX10003002);国家自然科学基金项目(31270176,81471910);国家杰出青年科学基金(81025008)
宋能,男,硕士在读,研究方向:感染与分子免疫,E-mail:Soner2008@qq.com;章晓联,通信作者,女,博士,教授,博士生导师,研究方向:病原微生物与感染,E-mail:zhangxiaolian@whu.edu.cn。
R378.911
A
1005-1678(2016)02-0010-06
猜你喜欢
人人健康(2022年13期)2022-07-25 07:14:30
电子元件与材料(2022年1期)2022-02-14 02:55:34
检验医学与临床(2022年1期)2022-01-26 07:25:12
郑州大学学报(工学版)(2022年1期)2022-01-17 01:58:16
河北医学(2021年10期)2021-10-27 00:37:14
纺织学报(2021年7期)2021-07-26 10:04:56
中国光学(2019年4期)2019-09-02 07:46:46
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
兽医导刊(2016年6期)2016-05-17 03:50:14
发明与创新(2015年25期)2015-02-27 10:39:16
