
CO2 methanation over TiO2-Al2O3 binary oxides supported Ru catalysts☆
2016-05-29 10:58JinghuaXuQingquanLinXiongSuHongminDuanHaoranGengYanqiangHuang
Jinghua Xu ,Qingquan Lin ,Xiong Su ,Hongmin Duan ,Haoran Geng *,Yanqiang Huang ,*
1 School of Materials Science and Engineering,Shandong University,Jinan 250061,China
2 State Key Laboratory of Catalysis,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,China
3 School of Chemistry and Chemical Engineering,Yantai University,Shandong Applied Research Center of Gold Nanotechnology(Au-SDARC),Yantai 264005,China
1.Introduction
The concentration of CO2in atmosphere continues to increase with the expansion of the world's population and economic growth.Therefore,areduction in CO2emission into the atmosphere is an urgent necessity.Among all the technologies(capture,sequestration,conversion,etc.),converting CO2into CH4has received renewed interests due to its us age as chemical storage of the excess H2generated from renew ableenergy and a solution for carbon resources recycling[1-3].Moreover,this reaction is regarded as a key technology for enabling longer manned space missions in the future,which is because of the recycling of wasted CO2from breathing and wasted H2from water electrolysis in the International Space Station[4-6].
The CO2methanation reaction is know n for a long time and was first introduced by Sabatier and Senderens[7],as the following reaction(Eq.(1)).

Notice that the reaction of CO2hydrogenation is strongly exothermic,albeit thermodynamically favored at low temperatures.Thus,the catalyst used in CO2hydrogenation must be efficient with a high activity at low temperatures and stable enough to endure the reaction heat.A series of VIIIB metals catalysts(Fe,Ru,Co,Rh,Ni)have been carried out on various supports(e.g.,SiO2,TiO2,Al2O3,ZrO2,CeO2)for the CO/CO2methanation reaction[8-13].Among all the investigated catalysts,Ru/TiO2has been regarded as the most efficient catalyst and has the highest selectivity towards CH4[14-17].It is reported that much higher activity on Ru/TiO2catalyst in the reaction of CO2methanation is resulted from a strong interaction between Ru and the TiO2support[18,19].Many works have been done to investigate the effects of different types of TiO2on catalytic activities in CO2methanation reaction,such as different facets[20],crystalline phases and morphologies of TiO2[21].Although TiO2has been regarded as the most effective support for Ru catalyst in CO2methanation,it presents the disadvantages of low surface area and poor structural stability w hen submitted to high temperature.Besides TiO2,another widely tested support for CO2methanation reaction is Al2O3,because it presents high surface area and resists to abrasion.How ever,Al2O3supported catalysts exhibited much lower catalytic activity than that of TiO2supported systems[22].
Incorporation of titania to alumina for preparation of titania-alumina compositeis now commonly used where great enhancement of catalytic activity is achieved as w ell as the higher surface area and thermal stability of titania-alumina mixture compared to the titania alone.TiO2-Al2O3binary oxides are widely applied as catalyst support[23].For instance,Lu et al.[24]have investigated the catalytic activities of Ru supported on TiO2-Al2O3binary oxide for the CO2methanation reaction.The results show ed that their catalytic activities were influenced by the content of TiO2in the supports.How ever,the influence of TiO2phase and the particle size of Ru on CO2conversions have not been reported in the literature.In this work,TiO2modified Al2O3binary oxide was prepared by using a w et-impregnation method,and used as the support for ruthenium catalyst(structure model is show n in Fig.1).The catalytic performance of Ru/TiO2-Al2O3catalyst in CO2methanation reaction was investigated.The effect of TiO2content and TiO2-Al2O3calcination temperature on the catalytic performance was addressed.

Fig.1.Structure model of TiO2-Al2O3 binary oxides supported Ru catalysts.
2.Experimental
2.1.Chemicals
Alumina(Al2O3,AR,SBET≥260 m2·g-1)was purchased from Aluminum Corporation of Shandong.Titanium tetra chloride(TiCl4,≥98.0%)and Ruthenium(III)chloride(RuCl3,11.1 wt%)solution were purchased from Sinopharm Chemical Reagent Co.,Ltd.Deionized water was used in all experiments.
2.2.Catalyst Preparation
2.2.1.Preparation of the TiO2-Al2O3 binary oxide
TiO2-Al2O3binary oxides with different mass concentrations of TiO2(5%,10%,15%)were prepared by a wet-impregnation method using Al2O3as the support.Typically,3 g Al2O3was added into the solution of TiCl4(10 vol%),and kept static at room temperature until the water was evaporated.Then,the samples were dried at 120°C for 12 h,and calcined in air at different temperatures(600,800,950,1100°C)for 4 h.
2.2.2.Preparation of the ruthenium catalysts
Al2O3or TiO2-Al2O3binary oxides supported Ru catalysts were prepared by incipient-wetness impregnation method with 5 w t%of metal loading.An aqueous solution(0.47 g)of RuCl3(11.1 w t%)was diluted to 0.85 g using ultra-super water.The Al2O3or TiO2-Al2O3support(1.00 g)was added into the solution and kept still at room temperature for 8 h.The obtained samples were dried at 120 °C for 12 h,then calcined in air at 300 °C for 4 h for further catalytic evaluations.
2.3.Activity test
The catalytic performance of these Ru catalysts in the CO2methanation reaction was carried out in a continuous- flow fixed-bed reactor under atmospheric pressure.50 mg catalyst(0.42-0.85 mm),diluted in 400 mg SiO2(0.42-0.85 mm),was loaded in a U-shape quartz reactor(i.d.6 mm).Before starting the reaction,the catalysts were reduced at 400°C for 1 h in the feed gas containing 18 vol%CO2,72 vol%H2and balanced with 10 vol%N2(employed as internal standard gas)with a flow rate of 50 ml·min-1.
The gas products from the reactor were passed through an ice-bath unit to remove the water vapor,and then analyzed on line with an Agilent 6890 gas chromatograph with a TDX-01 column connected to a TCD detector.The catalytic performance was expressed by the conversion of CO2based on different concentrations between inlet and outlet,which is defined as:
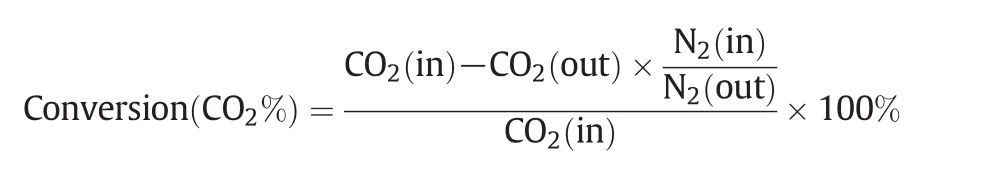
w here,CO2(in)and N2(in)are the CO2and N2concentrations at inlet;CO2(out)and N2(out)are the CO2and N2concentrations at outlet,respectively.We did not detect any CO or other by-products except CH4w hen the reaction temperatures were low er than 350°C.
2.4.Catalyst characterizations
The X-ray diffraction(XRD)patterns were recorded with a PANalytical X'Pert-Pro pow der X-ray diffractometer,using Cu Kαmonochromatized radiation(λ=0.1541 nm)operated at 40 kV and 40 m A.The scanning angle(2θ)range was from 10°to 90°,and operated with a scanning speed of 10(°)·min-1.
Temperature-programmed reduction(TPR)experiments were carried out on a Micromeritics AutoChem II2920 Automated Catalyst Characterization System.Prior to the H2-TPR experiment,about 50 mg of the calcined sample was loaded into a U-shape quartz reactor and pretreated with air at 150°C for 1 h to remove the adsorbed water.After cooling to room temperature,10 vol%H2/Ar mixed gas was passed through the sample and then heated to 900°C at a ramping rate of 10 °C·min-1.
Nitrogen adsorption-desorption isotherms were obtained on a Quadrasorb SI-20 apparatus at-196°C.Prior to the measurements,the samples were degassed at 300°C for 5 h.The Brunauer-Emmett-Teller(BET)method was used to calculate the specific surface areas(SBET).The pore size(Dp)and pore volume(Vp)were derived from the desorption branches of the isotherms using the Barrett-Joyner-Halenda(BJH)method.
High angular annular dark field scanning transmission electron microscopy(HAADF-STEM)images were recorded using a JEM-2100F field emission electronic microscope equipped with an STEM darkfield(DF)detector under the accelerating voltage of 200 kV.
3.Results and Discussion
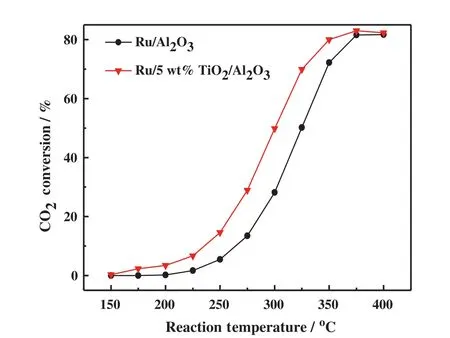
Fig.2.CO2 conversions as a function of the reaction temperature for Ru/Al2O3 and Ru/5 wt%TiO2-Al2O3 catalyst.TiO2-Al2O3 support was calcined at 950°C.
Fig.2 show s the catalytic activities as a function of reaction temperature over Ru/Al2O3and Ru/TiO2-Al2O3catalysts in the temperature range of 150-400°C.It is observed that the Ru/TiO2-Al2O3catalyst exhibited much higher activity than Ru/Al2O3.At the temperature of 225°C,about 6.7%CO2in the feed gas was converted to CH4on the Ru/TiO2-Al2O3catalyst,but the CO2conversion over Ru/Al2O3catalyst was only 1.7%.The reaction rates of the investigated catalysts at 225°C were calculated.There action rate on the Ru/TiO2-Al2O3catalyst was0.59 mol CO2·(g Ru)-1·h-1,3.1 times higher than that on Ru/Al2O3[0.19 mol CO2·(g Ru)-1·h-1],indicating that the TiO2-Al2O3binary oxide is a superior support to load Ru for CO2hydrogenation to CH4.
Fig.3 shows the XRD patterns of supported Ru catalysts on Al2O3and TiO2-Al2O3supports be fore and after reduction at 400°C.It can be clearly seen that the peak intensity of Ru O2on TiO2-Al2O3support before reduction is much weaker than that on Al2O3.Using Scherrer's equation,we get the crystal size of RuO2which is 8.3 nm when supported on Al2O3,while the peak intensity of Ru O2supported on TiO2-Al2O3is too weak to calculate the particle size.The relatively wide peaks indicate the small crystal size,suggesting that coating TiO2on the surface of Al2O3can effectively prohibit the aggregation of Ru O2.After the catalysts were reduced at 400°C for 1 h,the peak intensity of Ru supported on TiO2-Al2O3support is also much weaker than that on Al2O3,indicating the smaller particle size of Ru on TiO2-Al2O3support.
Fig.4 show s the STEM-HAADF images of supported Ru catalysts on Al2O3and TiO2-Al2O3support after reduction at 400°C for 1 h.We can easily find out that the Ru nanoparticles(displayed as bright round spots in the STEM-HAADF images)on the surface of Al2O3support are larger than those on TiO2-Al2O3.The averaged particle size of the Ru on Al2O3support is found to be(4.3±2.5)nm.How ever,it is only about(2.8±1.6)nm and with a narrower particle size distribution for the Ru/TiO2-Al2O3catalyst.CO chemisorption on the catalysts was also carried out to determine the dispersion of Ru metal.Results show ed that the dispersion of Ru on Ru/Al2O3is about 4.7%,but this value was increased to 5.5%on the Ru/TiO2-Al2O3catalyst.We further calculated the turnover frequency(TOF)of CO2conversion with respect to the number of surface Ru atoms at 225°C.The TOF over Ru/Al2O3and Ru/TiO2-Al2O3catalysts are 0.11 and 0.30 s-1,respectively.These results indicate that coating TiO2on the surface of Al2O3appears to be an efficient strategy to stabilize smaller nano-sized particles of Ru by a strong interaction between Ru and the TiO2support.It is considered as one of the reasons for the enhanced activity.
The influence of TiO2loading on CO2conversions for supported Ru catalysts is studied.Fig.5 displays the catalytic activities as a function of reaction temperature over supported Ru catalysts with different loadings of TiO2.We can see that it has little impact on the catalytic activities when the TiO2loading increases from 5 wt%to 15 w t%.XRD patterns of the corresponding catalysts are show n in Fig.6.It is interesting to find out that there exist two structure types of Al2O3(α-Al2O3and θ-Al2O3)in these catalysts.The peak intensity of α-Al2O3is enhanced gradually with the increasing loading of TiO2,indicating that the phase transformation of θ-Al2O3to α-Al2O3in TiO2-Al2O3complex support can be promoted as the loading of TiO2increases.At the same time,the BET surface area of TiO2-Al2O3decreases from 67 to 56 m2·g-1(see Table 1)along with the θ-Al2O3to α-Al2O3phase transformation.In addition,the peak intensity of r-TiO2is also enhanced with the increasing loading of TiO2,indicating that the particle size of r-TiO2becomes bigger.The bigger particle size of r-TiO2and decreased specific surface area of TiO2-Al2O3along with the increasing loading of TiO2may be the possible reasons for the unimproved activity.
Fig.7 displays the catalytic activities over supported Ru catalysts as the calcination temperature of TiO2-Al2O3binary oxide support increased from 600 to 1100°C.Obviously,the CO2conversion increases with the increase of calcination temperature(from 600 to 950°C).However,further increase the calcination temperature to 1100°C,the CO2conversion doesn't increase.On the contrary,it shows a decreased tendency within the reaction temperature range of 150-350°C.The CO2conversion increases only w hen the reaction temperature is above 350°C,which may be attributed to a different reaction mechanism to be further identified.From the above discussions,the optimal calcination temperature of the support is considered to be 950°C.
To investigate the relation ship between catalytic activities and structural properties of the catalysts after calcination at different temperatures,the powdered X-ray diffraction measurements were performed.From the results(Fig.8),it can be seen that the signals of anatase-TiO2(abbreviated as a-TiO2)disappears w hen the calcination temperature of TiO2-Al2O3binary oxide increases to 950°C,which is considered to be transformed to rutile phase.How ever,the relative XRD peak intensity is too weak to be identified due to the small crystal size of rutile.At the same time,γ-Al2O3is transformed to θ-Al2O3,leading to the specific surface area of TiO2-Al2O3decrease from 181 to 67 m2·g-1(Table 1).When the calcination temperature further increases to 1100°C,the rutile crystal signals could be clearly observed and its intensity greatly enhanced,indicating the bigger particle size of the support.Meanwhile,θ-Al2O3is transformed to α-Al2O3,and the corresponding specific surface area of TiO2-Al2O3decreases to 47 m2·g-1(Table 1).Both of the factors would lead to the aggregation of metal particles with the loss of surface active sites,which should be responsible for the decreased conversion of CO2.It is previously reported that in Al2O3-TiO2complex support,the presence of TiO2could reduce the phase transformation temperature of Al2O3.How ever,the presence of Al2O3could raise the temperature of anatase to rutile TiO2transformation[25,26].In addition,the peak intensity of Ru O2weakened along with the anatase to rutile TiO2transformation,indicating that RuO2particle size is decreased.Using Scherrer's equation,w e obtained that the crystal size of Ru O2were 8.2 nm and 7.4 nm w hen the Al2O3-TiO2binary support was calcined at 600 and 800°C,respectively.How ever,w hen the calcination temperature of the support is above 950°C,the peak intensity of Ru O2is too weak to calculate the particle size.Therefore,by analyzing the XRD patterns of these catalysts,we conclude that r-TiO2appears to be a better support than a-TiO2to improve the dispersion of RuO2.This is consistent with a previous report that there was a strong interaction between Ru O2and r-TiO2,which prohibited the aggregation of RuO2[19].As a result,the smaller particle size of Ru on TiO2-Al2O3support is obtained,which is considered to be one of the reasons for the improved activity in CO2methanation.
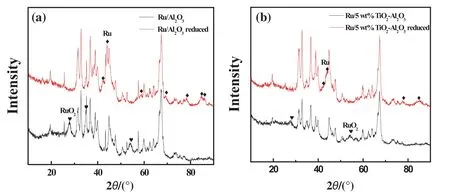
Fig.3.XRD patterns of supported Ru catalysts.(a)Ru/Al2O3 and(b)Ru/5 wt%TiO2-Al2O3.TiO2-Al2O3 support was calcined at 950°C.
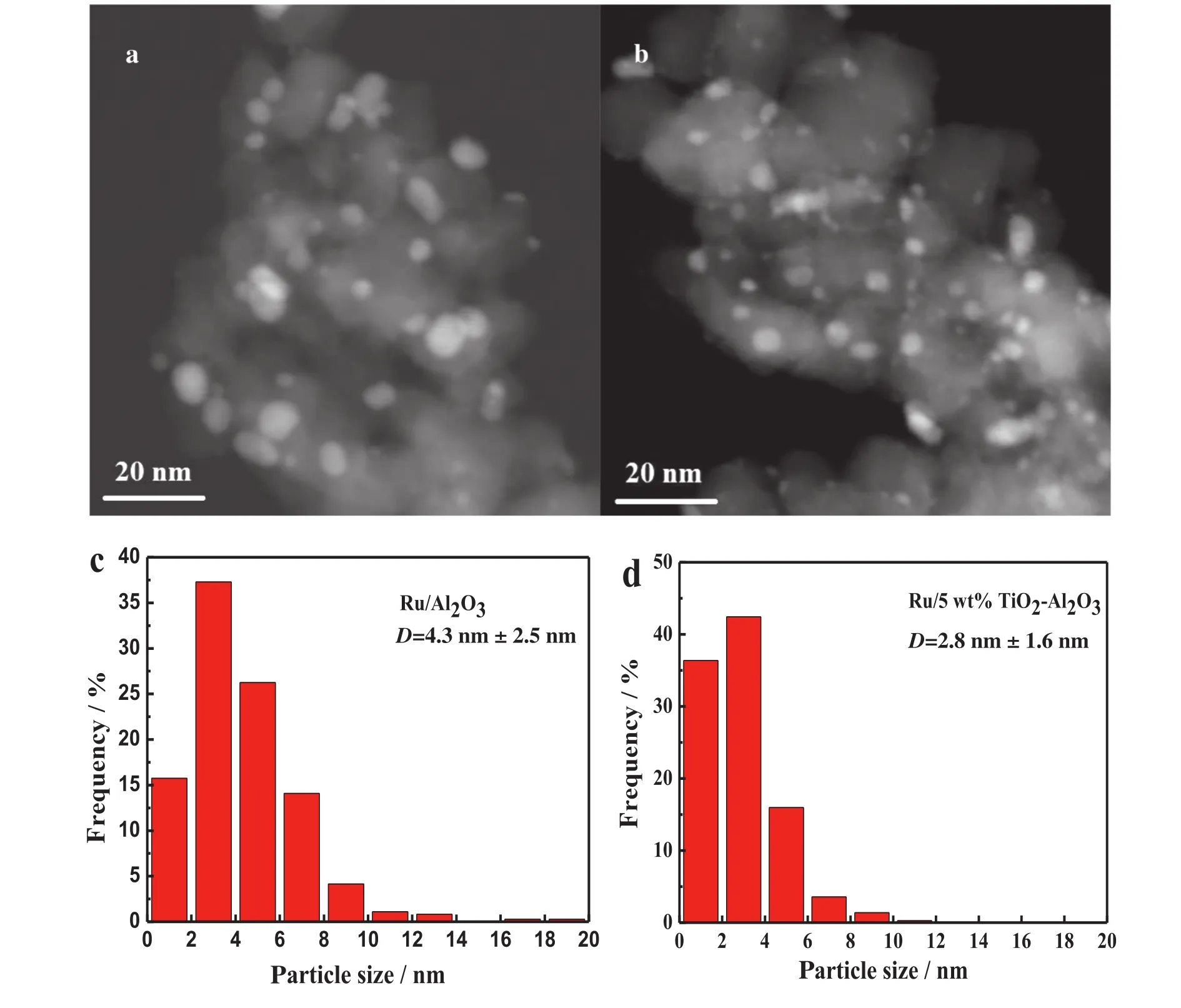
Fig.4.STEM-HAADF images and size distribution of supported Ru catalysts(a)&(c)Ru/Al2O3;(b)&(d)Ru/5 wt%TiO2-Al2O3.TiO2-Al2O3 support was calcined at 950°C.
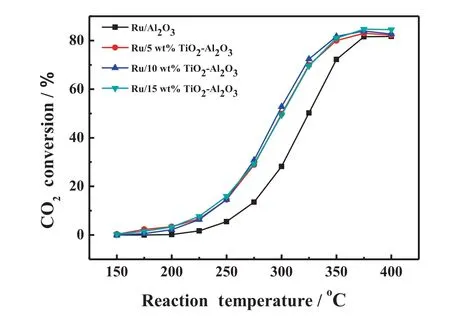
Fig.5.Effect of TiO2 loading on CO2 conversion for supported Ru catalysts.TiO2-Al2O3 support was calcined at 950°C.
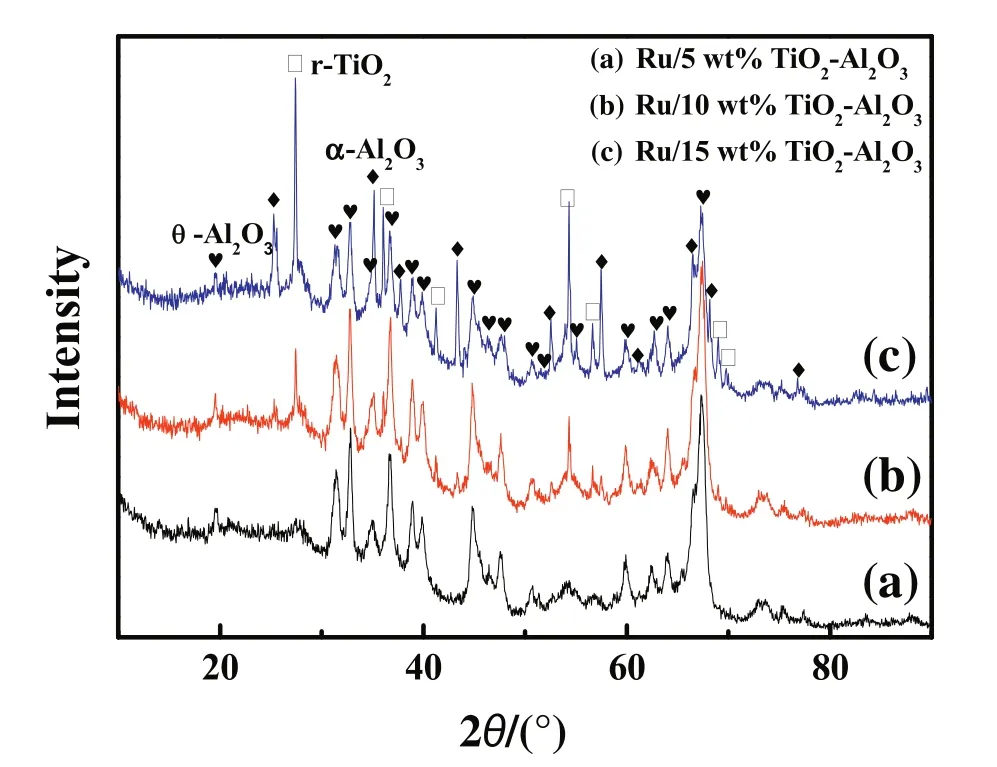
Fig.6.XRD patterns of supported Ru catalysts with different TiO2 loading.TiO2-Al2O3 support was calcined at 950°C.
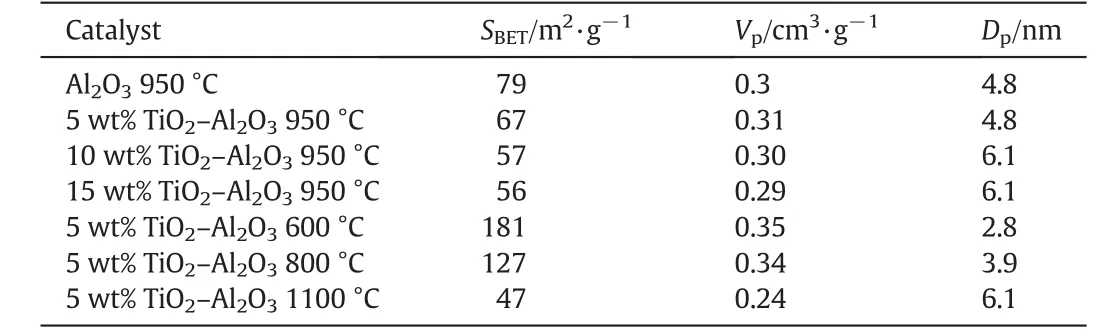
Table 1 Surface measurements for TiO2-Al2O3 supported catalysts

Fig.7.Effect of calcination temperatures of supports on CO2 conversions for supported Ru catalysts.
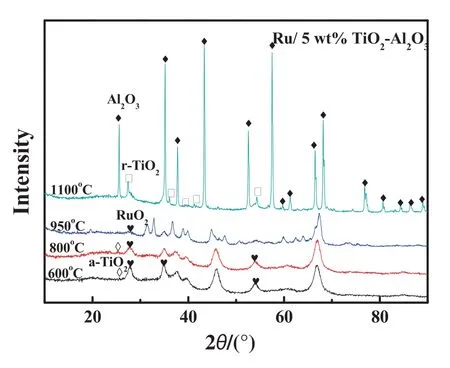
Fig.8.XRD patterns of the supported Ru catalysts at different calcination temperatures.
Fig.9 show s H2-TPR pro files of the supported Ru catalysts on TiO2-Al2O3binary oxide which are calcined at different temperatures.The Ru O2species start to be reduced at 156°C w hen the calcination temperature is 600°C,but the reduction temperature is delayed to 162 °C w hen the calcination temperature increases to 800 and 950 °C.The most likely explanation is the strong interaction between RuO2and r-TiO2,which hindered the reduction of RuO2.How ever,the reduction temperature of RuO2is reduced to 146°C when the calcination temperature further increases to 1100°C,which is probably due to the weakened interaction between Ru O2and r-TiO2originated from the much larger particle size of r-TiO2.
By analyzing these results,w e get the know ledge that the presence of TiO2in Al2O3-TiO2complex support facilitated the phase transformation of Al2O3,leading to a decrease of the specific surface area of Al2O3-TiO2complex support.Therefore,grafting titanium on other thermally stable supports,such as SiO2,seems to be a better option to prepare the binary oxide support.That part of research is under investigation in our research group.The results will be reported separately later on.
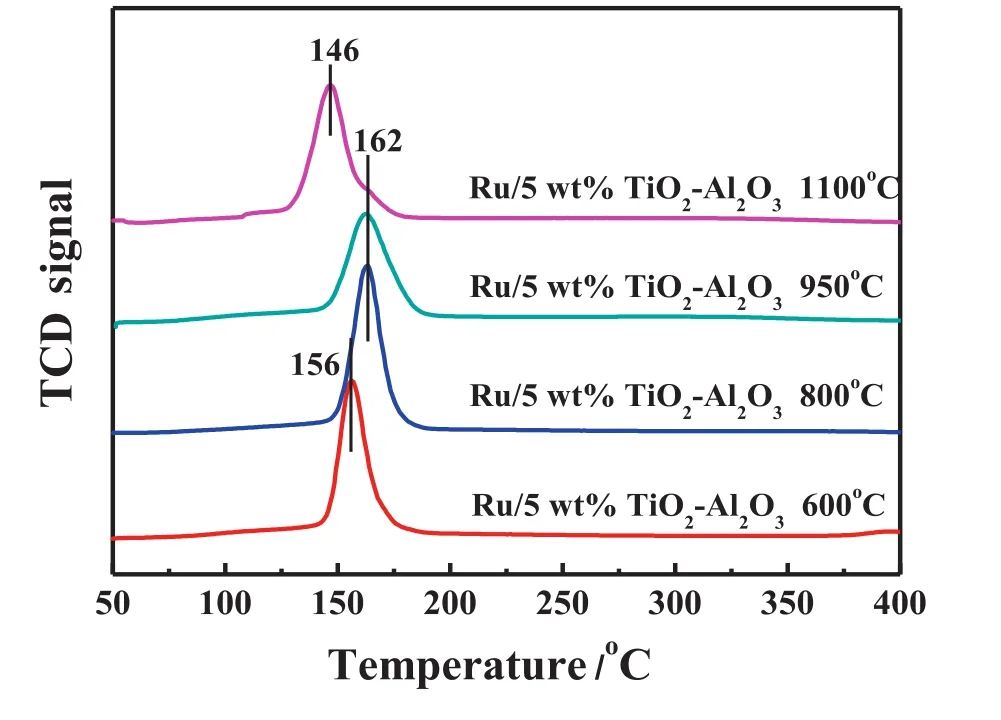
Fig.9.H2-TPR pro files of the supported Ru catalysts at different calcination temperatures.
4.Conclusions
In this study,TiO2modified Al2O3binary oxide was prepared by a w et-impregnation method and used as a support for ruthenium catalyst.The catalytic performance of Ru/TiO2-Al2O3catalyst in CO2methanation reaction was tested.The Ru/TiO2-Al2O3catalyst exhibited a much higher activity in CO2methanation reaction than the Ru/Al2O3catalyst.The reaction rate on the Ru/TiO2-Al2O3catalyst was 0.59 mol CO2·(g Ru)-1·h-1,3.1 times higher than that on Ru/Al2O3[0.19 mol CO2·(g Ru)-1·h-1].The effect of TiO2content and TiO2-Al2O3calcination temperature on catalytic activity was addressed.It was found that TiO2content has little impact on the catalytic activity in CO2methanation reaction.But the phase of TiO2has a great influence on activity of catalysts.When the calcination temperature of TiO2-Al2O3increased from 600 to 950°C,anatase TiO2transformed into rutile phase,resulting in smaller Ru particle size(2.8 nm)than that on Al2O3(4.3 nm).The small Ru particle size,originated from a strong interaction between Ru and the r-TiO2support,should be responsible for the improved activity over Ru/TiO2-Al2O3catalyst.
[1]W.Wang,S.P.Wang,X.B.Ma,J.L.Gong,Recent advances in catalytic hydrogenation of carbon dioxide,Chem.Soc.Rev.40(7)(2011)3703-3727.
[2]D.J.Cheng,F.R.Negreiros,E.Aprà,A.Fortunelli,Computational approaches to the chemical conversion of carbon dioxide,ChemSusChem 6(6)(2013)944-965.
[3]L.He,Q.Q.Lin,Y.Liu,Y.Q.Huang,Unique catalysis of Ni-Al hydrotalcite derived catalyst in CO2methanation:Cooperative effect between Ni nanoparticles and a basic support,J.Energy Chem.23(5)(2014)587-592.
[4]J.X.Liu,W.H.Hou,Study on Ru-based catalyst used in reductive reaction of CO2,Space Med.Med.Eng.17(6)(2004)457-460(in Chinese).
[5]Y.Y.Meng,C.X.Shang,A study on CO2methanization reduction technology,Space Med.Med.Eng.7(2)(1994)115-120(in Chinese).
[6]K.H.Zhou,B.Z.Wu,C.B.Ren,Comparative analysis of Sabatier CO2reduction system for space station,Space Med.Med.Eng.24(5)(2011)384-390(in Chinese).
[7]P.Sabatier,J.B.Senderens,New synthesis of methane,C.R.Acad.Sci.134(1902)514-516.
[8]Q.H.Liu,X.F.Dong,Z.L.Liu,Performance of Ni/Nano-Zr O2catalysts for CO preferential methanation,Chin.J.Chem.Eng.22(2)(2014)131-135.
[9]I.Graca,L.V.González,M.C.Bacariza,A.Fernandes,C.Henriques,CO2hydrogenation into CH4on NiHNaUSY zeolites,Appl.Catal.B Environ.147(2014)101-110.
[10]W.Wang,J.L.Gong,Methanation of carbon dioxide:an overview,Front.Chem.Sci.Eng.5(1)(2011)2-10.
[11]G.D.Weatherbee,C.H.Bartholomew,Hydrogenation of CO2on group VIII metals:IV.Specific activities and selectivities of silica-supported Co,Fe,and Ru,J.Catal.87(2)(1984)352-362.
[12]Q.H.Liu,X.F.Dong,Z.L.Liu,Performance of Ni/nano-ZrO2catalysts for CO preferential methanation,Chin.J.Chem.Eng.22(2)(2014)131-135.
[13]J.Liu,D.M.Cui,J.Yu,F.B.Su,G.W.Xu,Performance characteristics of fluidized bed syngas methanation over Ni-Mg/Al2O3catalyst,Chin.J.Chem.Eng.23(1)(2015)86-92.
[14]J.A.Mieth,J.A.Schwarz,The effect of catalyst preparation on the performance of alumina-supported ruthenium catalysts:I.The impact of catalytic precursor on particle size and catalytic activity,J.Catal.118(1)(1989)203-217.
[15]Q.Jiang,G.C.Deng,R.T.Chen,Z.T.Huang,A study on catalysts for methanation of carbon dioxide I.The activity of supported group VII metal catalysts,Chin.J.Catal.18(1)(1997)5-8.
[16]S.Sharma,Z.Hu,P.Zhang,E.W.McFarland,H.Metiu,CO2methanation on Ru-doped ceria,J.Catal.278(2)(2011)297-309.
[17]D.Li,N.Ichikuni,S.Shimazu,T.Uematsu,Hydrogenation of CO2over sprayed Ru/TiO2fine particles and strong metal-support interaction,Appl.Catal.A 180(1-2)(1999)227-235.
[18]C.M.Li,S.T.Zhang,B.S.Zhang,D.S.Su,S.He,Y.F.Zhao,J.Liu,F.Wang,M.Wei,D.G.Evans,X.Duan,Photohole-oxidation-assisted anchoring of ultra-small Ru clusters onto TiO2with excellent catalytic activity and stability,J.Mater.Chem.A 1(7)(2013)2461-2467.
[19]Q.Q.Lin,X.Y.Liu,Y.Jiang,Y.Wang,Y.Q.Huang,T.Zhang,Crystal phase effects on the structure and performance of ruthenium nanoparticlesfor CO2hydrogenation,Catal.Sci.Technol.4(7)(2014)2058-2063.
[20]F.Wang,S.T.Zhang,C.M.Li,J.Liu,S.He,Y.F.Zhao,H.Yan,M.Wei,D.G.Evans,X.Duan,Catalytic behavior of supported Ru nanoparticles on the(101)and(001)facets of anatase TiO2,RSC Adv.4(21)(2014)10834-10840.
[21]G.Y.Wang,Y.X.Gao,W.D.Wang,W.X.Huang,Selective CO methanation over Ru catalysts supported on nanostructured TiO2with different crystalline phases and morphology,Chin.J.Chem.Phys.25(4)(2012)475-480.
[22]M.H.Brijaldo,F.B.Passos,H.A.Rojas,P.Reyes,Hydrogenation of m-dinitrobenzene over Pt supported catalysts on TiO2-Al2O3binary oxides,Catal.Lett.144(5)(2014)860-866.
[23]A.E.Awadallaha,M.S.Mostafa,A.A.Aboul-Enein,S.A.Hana fib,Hydrogen production via methane decomposition over Al2O3-TiO2binary oxides supported Ni catalysts:effect of Ti content on the catalytic efficiency,Fuel 129(2014)68-77.
[24]H.X.Lu,B.H.Qin,K.P.Sun,X.L.Xu,Study of Ru/Al2O3-TiO2catalyst for CO2methanation,J.Mol.Catal.19(1)(2005)27-30(in Chinese).
[25]S.Nakade,M.Matsuda,S.Kambe,Y.Saito,T.Kitamura,T.Sakata,Y.Wada,H.Mori,S.Yanagida,Dependence of TiO2nanoparticle preparation methods and annealing temp erature on the efficiency of dye-sensitized solar cells,J.Phys.Chem.B 106(39)(2002)10004-10010.
[26]Y.Wei,X.X.Liu,Preparation and characterization of Al2O3-TiO2complex support,Petrochem.Technol.35(2)(2006)173-177(in Chinese).
 Chinese Journal of Chemical Engineering2016年1期
Chinese Journal of Chemical Engineering2016年1期
- Chinese Journal of Chemical Engineering的其它文章
- Scoping biology-inspired chemical engineering☆
- Review on the nanoparticle fluidization science and technology☆
- Multi-functional forward osmosis draw solutes for seawater desalination☆
- Bio-inspired enantioseparation for chiral compounds☆
- Process engineering in electrochemical energy devices innovation☆
- In-situ design and construction of lithium-ion battery electrodeson metal substrates with enhanced performances:A brief review☆