
拉伸条件下Cu100-xNix局部结构演化分子动力学研究*
2016-05-27 09:30:57梁红玉
新技术新工艺 2016年4期
李 烨,吕 明,梁红玉
(1.太原工业学院 机械工程系,山西 太原 030008;2.太原理工大学 机械工程学院,山西 太原 030051)
拉伸条件下Cu100-xNix局部结构演化分子动力学研究*
李烨1,2,吕明2,梁红玉1
(1.太原工业学院 机械工程系,山西 太原 030008;2.太原理工大学 机械工程学院,山西 太原 030051)
摘要:利用分子动力学模拟研究Cu100-xNix(x=5,10和15)纳米杆单向拉伸过程,比较分析了不同Ni含量对应力-应变关系的影响,并且应用HA键对指数方法对重要应变处的结构进行了分析。模拟结果显示,Ni含量增加对最大应力影响不大,其影响主要发生在拉伸后期,使应力下降更加迅速。拉伸过程中,主要的键对指数是1441、1661和1551。1551指数含量减少使得1441和1661指数含量升高,导致了应力值升高;反之亦然。Ni含量越大,对于成键总量变化影响也越大。
关键词:分子动力学;CuNi合金;拉伸;HA键对指数分析
CuNi合金具有良好的室温力学性能和高温强度[1],耐蚀性高耐磨性好[2],得到了广泛深入应用。然而,在纳米尺度下合金变形试验研究受到了较大制约,主要表现在变形条件加载控制以及变形过程中的检测;因此,尽管试验研究已经有了一些进展,但是对于纳米级的二元合金拉伸过程中的结构演化仍然缺乏较为清晰的认识。
分子动力学方法作为研究复杂凝聚态系统的强有力工具,在金属和合金非晶态结构和性质研究中取得了许多成果。目前,许多研究者将分子动力学应用于金属纳米材料变形行为的研究。如V. S. Krasnikov等模拟在不同温度和不同尺寸空穴下对单晶Al的拉伸力学性能的影响,发现温度的提高减小了其拉伸强度,而减小空穴尺寸却增加了强度[3]。 J. Ren等通过模拟单晶Ti纳米线拉伸,发现了较为罕见的HCP到FCC的相变,并且指出这种相变是由Shockley不全位错引起[4]。G. Sainath等通过对Fe纳米线不同横截面尺寸的拉伸行为进行模拟时,发现了横截面尺寸对弹性模量、屈服强度和流动应力的影响规律[5]。P. H. Sung等在研究Ni金属裂隙生长和传播模拟时,指出低温时容易发生脆性断裂,并且拉伸时晶向位置决定了极限应力的大小[6]。
当前,分子动力学研究主要集中在纯金属等方面,对于二元合金体系的报道相对较少。本试验以低含量Ni的Cu100-xNix(x=5,10和15)二元合金作为模型,采用分子动力学模拟该合金在纳米尺度下的拉伸过程,采用键值对数方法(HA)分析了拉伸过程中合金结构的演化过程。
1模拟方法和过程
本次试验使用分子动力学对Cu100-xNix在纳米尺度下的拉伸行为进行模拟,并且用HA对其结构演化进行分析。试验中采用Finnis-Sinclair EAM(FSEAM)[7]作用势对Cu-Ni原子间关系进行表述。在FSEAM势中,晶体总势能E由位于晶格点阵上的原子核之间的对势能,以及原子核镶嵌在电子云背景中的嵌入能两部分组成:
式中,ρα是α类型元素的嵌入能函数;fαβ是两元素的电子云密度函数;γij是原子i和原子j之间的距离;φαβ是两元素之间的对势函数。文中采用的势能函数来源于文献[8],此处不再详细叙述。
采用LAMMPS软件包对Cu100-xNix二元合金纳米杆拉伸行为进行了模拟。模型初始结构为长方体,其尺寸约为10.83 nm×3.61 nm×3.61 nm(见图1)。晶格结构采用FCC结构,x方向对应[100]晶向,y方向对应[010]方向,z方向对应[001]方向,共含有12 000个原子。Ni原子为置换元素,按照5at%、10at%和15at%的比例随机置换Cu原子。模拟时,首先在300 K温度下,弛豫50 000步,最终达到稳定,步长设定为0.001 ps;然后在300 K温度和应变速率0.01 ps-1条件下拉伸20 ps;最后用模拟数据求得应力-应变关系,探讨了不同Ni含量对Cu100-xNix二元合金拉伸行为的影响,并使用HA方法对拉伸过程中特定应变处进行了键对指数分析。
图1 Cu85Ni15拉伸模型
2结果与讨论
Cu95Ni5在温度为300 K,应变速率为0.01 ps-1,拉伸20 ps时的应力-应变曲线如图2所示。在图2中,各个曲线可分为2个阶段:1)线性阶段,应力值大体呈线性增加,与宏观材料的弹性变形阶段相似,其现象说明纳米尺度下仍然符合胡克定律;2)应力值波动阶段,应力值在最大应力之后由于拉伸过程中不断有金属键断裂使得应力值下降,同时原子移位后又与相邻原子形成新的金属键,应力值再次上升。由于金属键断裂的程度大于新金属键形成的程度,导致了这一阶段应力-应变曲线呈波动下降的现象。当应变ε为0.15时,应力达到最大值(约为15.6 GPa)。
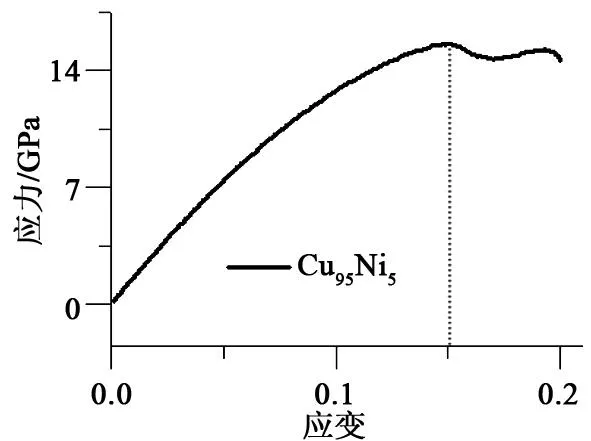
图2 二元Cu95Ni5合金应力-应变曲线
Cu100-xNix(x=5,10和15)在温度为300 K,应变速率为0.01 ps-1,拉伸20 ps时的应力-应变曲线如图3所示。从图3中可以看到,这3种合金在拉伸第1阶段基本相似,最大应力值虽然随着Ni含量增加在逐渐减小,但是减小幅度很小,这说明在弹性阶段Ni元素含量变化对合金拉伸行为的影响较小。第2阶段拉伸后期,Ni含量对拉伸行为影响较大,特别是Cu85Ni15,其原因可能是Ni含量增大使新金属键强度较低,从而应力迅速下降。为了更进一步理解Cu100-xNix拉伸过程中结构变化,研究中使用HA键对分析对二元合金在关键应变处(ε=0.0,0.15,0.17,0.20)进行了结构演化分析。
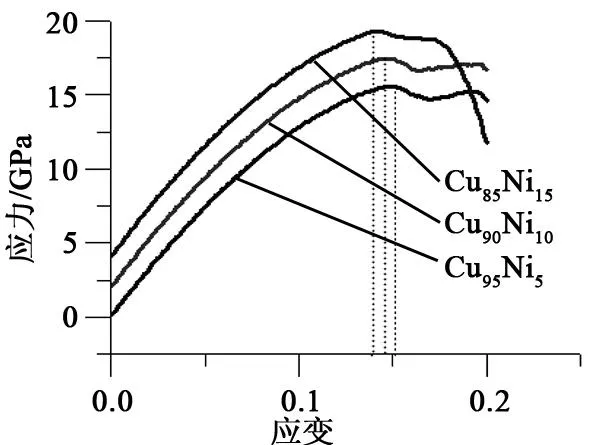
图3 二元Cu100-xNix合金应力-应变曲线
HA键对指数方法是描述非晶态和晶态体系原子结构的一种有效方法[9-11]。HA键对指数方法用4个指数ijkl来描述局部原子结构,一般而言1551、1541和1431指数大量存在于液态或非晶中。1441和 1661指数代表了bcc结构。 1421指数为fcc晶体结构的特征指数,hcp晶体结构存在特征指数是1421和1422。
Cu100-xNix(x=5,10和15)在拉伸过程中各种重要键对指数百分含量在应变(ε=0.0,0.15,0.17,0.20)处的分布如图4所示。3种不同Ni含量的合金在拉伸过程中,各种键对指数变化非常相似。模拟系统中主要的指数代表bcc结构(1441和1661)和非晶(1551),占到了总量的70%以上。当0<ε<0.15时,1441和1661含量逐渐增多,同时1551、1541和1431减少,尤其是1541和1431指数;当0.15<ε<0.17时,1441和1661含量逐渐增多,同时1551减少;当0.17<ε<0.2时,1441和1661含量逐渐减少,同时1551、1541和1431指数增多。已有的研究表明,1551、1541和1431指数在拉伸过程中有着重要的作用[12-14]。拉伸时首先是1551、1541和1431代表的结构变得无序和更加松弛,使得应力下降,然后转变为更为稳定的结构,又导致了应力上升。另外,Ni含量越大,对于成键总量变化影响也越大,如图4d所示,尤其是Cu85Ni15在ε=0.20时成键总量减少非常明显,使得应力迅速减少。
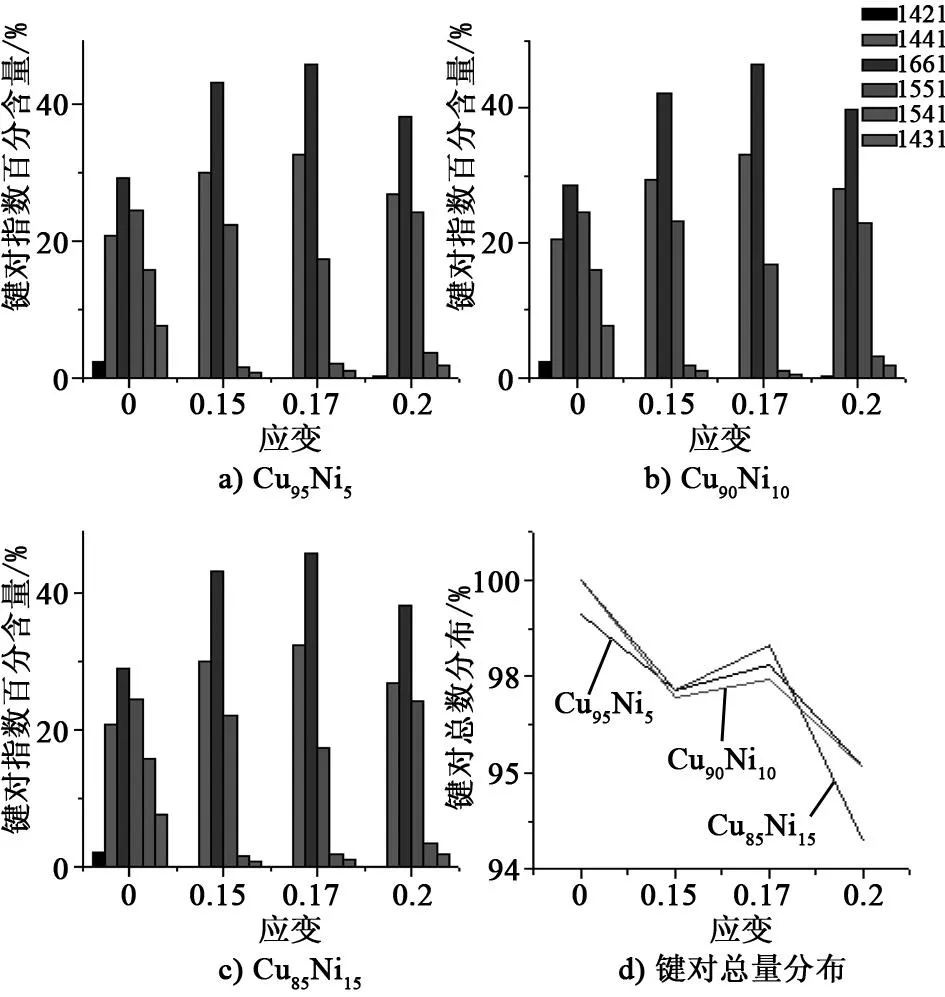
图4 Cu100-xNix在拉伸过程键对指数分布
3结语
Cu100-xNix(x=5,10和15)在模拟中的拉伸现象与宏观材料相似,经历了弹性阶段和随后的应力波动阶段。在弹性阶段Ni含量对最大应力影响程度不大;但是Ni含量对随后的应力波动有较大的影响,Ni含量越大,拉伸后期应力越小。在拉伸过程中,1441、1661和1551指数含量占到了成键总量的70%以上,并且随着1551指数含量减少,1441和1661指数增多,导致了应力值升高;反之亦然。
参考文献
[1] Qi Y, Augin T, Kimura Y. Molecular-dynamics simulations of glass formation and crystallization in binary liquid metals: Cu-Ag and Cu-Ni[J]. Phys Rev B, 1999,59(5):3527-3529.
[2] Fenelon A M, Breslin C B. The electropolymerization of pyrrole at a CuNi electrode: corrosion protection properties[J]. Corros Sci, 2003,45(12):2837-2850.
[3] Krasnikov V S, Mayer A E. Plasticity driven growth of nanovoids and strength of aluminum at high rate tension: Molecular dynamics simulations and continuum modeling[J]. Int J Plast, 2015,74:75-91.
[ 4] Ren J, Sun Q, Xiao L,et al. Phase transformation beha vior in titanium single-crystal nanopillars under [0 0 0 1] orientation tension: A molecular dynamics simulation[J]. Comput Mater Sci, 2014,92:8-12.
[5] Sainath G, Choudhary B K. Molecular dynamics simulations on size dependent tensile deformation behaviour of [110] oriented body centred cubic iron nanowires[J]. Mater Sci Eng A, 2015,640:98-105.
[6] Sung P H, Chen T C. Studies of crack growth and propagation of single-crystal nickel by molecular dynamics[J]. Comput Mater Sci, 2015,102:151-158.
[7] Rafii-Tabar H, Sulton A P. Long-range Finnis-Sinclair potentials for fcc metallic alloys[J]. Philos Mag Lett, 1991,63(4):217-224.
[8] Bonny G, Pasianot R C, Castin N, et al. Ternary Fe-Cu-Ni many-body potential to model reactor pressure vessel steels: First validation by simulated thermal annealing[J]. Philos Mag, 2009,89(34):3531-3546.
[9] Honeycutt J D, Andersen H C. The effect of periodic boundary conditions on homogeneous nucleation observed in computer simulations[J]. Chem Phys Lett, 1984;108(6):535-538.
[10] Honeycutt J D, Andersen H C. Small system size artifacts in the molecular dynamics simulation of homogeneous crystal nucleation in supercooled atomic liquids[J]. J Phys Chem, 1986,90(8):1585-1589.
[11] Honeycutt J D, Andemen H C. Molecular dynamics study of melting and freezing of small lennard- jones clusters[J]. J Phys Chem,1987,91:4950-4963.
[12] Huang D, Tao W, Guo Y. Molecular dynamics simulation of failure process of nano aluminum wire under axial tension [J]. Ordnance Mater Sci Eng, 2005(3):2.
[13] Wolf D, Yamakov V, Phillpot S R,et al. Deformation of nanocrystalline materials by molecular-dynamics simulation: relationship to experiments[J]. Acta Mater, 2005,53(1):1-40.
[14] Wakeda M, Shibutani Y, Ogata S,et al. Relationship between local geometrical factors and mechanical properties for Cu-Zr amorphous alloys[J]. Intermetallics, 2007,15(2):139-144.
责任编辑马彤

The Local Structural Evolution of Cu100-xNixduring Tensile Deformation: A Molecular Dynamics Study
LI Ye1,2, LYU Ming2, LIANG Hongyu1
(1.Department of Mechanical Engineering, Taiyuan Institute of Technology, Taiyuan 030008, China;2.College of Mechanical Engineering, Taiyuan University of Technology,Taiyuan 030024, China)
Abstract:The deformation of Cu100-xNix under uniaxial tension has been conducted by using molecular dynamics (MD) simulations. The influence of Ni content on the stress-strain curve is analyzed, and the Honeycutt-Andersen pair analysis (HA) is calculated to determine structure on the key point of strain-stress curves. The result shows that the Ni content does not have effect on the maximum stress, but it has effect on stress fluctuation at the end of simulation. The main bond pairs are 1441,1661 and 1551 during the tension simulation. The decrease of 1551 leds to the 1441 and 1661 increasing. The Ni contends has effect on the total number of boned pairs, which influences the stress.
Key words:molecular dynamics, CuNi alloy, uniaxial tension, Honeycutt-Andersen pair analysis
中图分类号:TG 146.2+3
文献标志码:A
收稿日期:2015-12-01
作者简介:李烨(1978-),男,讲师,博士,主要从事纳米材料及其力学性能等方面的研究。
* 山西国际科技合作项目(2013081021)
