
有机小分子催化不对称环氧化
2016-05-12 07:03周文利张焰臣居学成彭琴琴
海南师范大学学报(自然科学版) 2016年2期
马 龙,陈 寿,周文利,张焰臣,居学成,彭琴琴,郑 超,*
(1.海南师范大学 化学与化工学院,海南 海口 571158;2.深圳市赛欣瑞科技创新中心,广东 深圳 518110)
有机小分子催化不对称环氧化
马 龙1,陈 寿2,周文利1,张焰臣1,居学成2,彭琴琴1,郑 超1,2*
(1.海南师范大学 化学与化工学院,海南 海口 571158;2.深圳市赛欣瑞科技创新中心,广东 深圳 518110)
光学活性的环氧化合物是有机合成的重要原料.不对称环氧化反应可以使潜手性的烯烃转化为带有手性碳的环氧化合物,这个反应在医药、农药、香料等合成上具有重要的意义.有机小分子催化烯烃的不对称环氧化是一种高效,绿色的光学活性环氧化合物的合成方法,目前该类反应主要是通过手性酮和手性亚胺盐来催化实现的.文章介绍了该类反应的催化机理,并概述了这两类有机小分子催化的不对称环氧化反应的研究进展.
不对称环氧化;手性酮;手性亚胺正离子
1 手性酮催化碳碳双键的不对称环氧化
手性酮催化碳碳双键的不对称环氧化是通过手性二氧杂环丙烷中间体——Dioxirane进行的反应,Dioxirane可由酮和过硫酸氢钾复合盐(2KHSO5·KHSO4·K2SO4)原位生成[1].一般来说,只需一定催化量的酮即可以推进反应,因为当Dioxiranes活泼性的氧原子转移到碳碳双键上,会重新生成酮,催化循环得以不断进行下去[2].其催化循环机理见图1.
然而实际上手性酮催化剂的发展是一个很具有挑战性的问题,因为手性酮的位阻和电子效应在反应活性和对映选择性上的竞争是一个很大的难题.
1984年,Curci等人首次报道了用图2中酮1与酮2催化的不对称环氧化[3],酮的用量为0.2~3 eq,虽然可以取得较好的产率,但对映体过量百分率(ee值)最高值反为12.5%.之后在1995年,Curci等人又报道了催化剂酮3和酮4对反式二苯乙烯或反式甲基苯乙烯的环氧化[4],这两种酮的活性比前面提到的两种酮要高,酮的用量也降到0.8~1.2 eq,并且ee值上升到20%.
1996年,杨丹等人报道了一系列联萘基衍生的手性酮5(见图3),酮5是首次报道的可以获得具有实用价值的ee值的酮[5],催化剂的用量最小可以降到10 mol%,并发现在联萘基邻位的手性选择“X”基团对控制选择性环氧化起到很大的作用.在使用等摩尔量联萘基衍生的酮5a作为催化剂时[6],对反式-4,4’-二苯基-二苯乙烯的催化中ee值为87%.双缩醛5d是这一系列中反应活性和选择性最好的环氧化催化剂,在反式1,2-二苯乙烯的催化环氧化中,产率为93%,ee值为84%.
然而酮5a~d的合成需要相当昂贵的起始原料以及一些危险昂贵的反应才能制备,且一般催化剂用量较高(通常相对于底物摩尔数不少于10%),这些因素导致了它们难以得到广泛的应用.Denmark等人分别在1999年7月和2002年8月报道了手性七元环酮6.
与11元环酮5相比,七元环酮的反应活性更高了,当在酮羰基的邻位加入强吸电子基团氟来控制立体效应之后,酮6c和6d的反应活性比起6a来有较大幅度的提高.对1-苯基-丙烯的不对称环氧化产率80%,ee值达到88%.
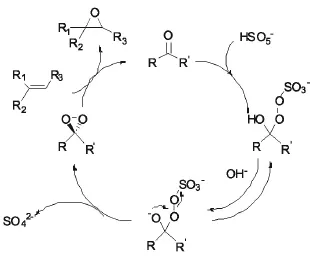
图1 酮催化烯烃环氧化Fig.1 Ketones catalyzed olefin epoxidation
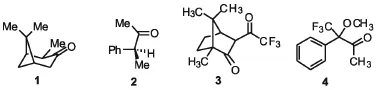
图2 最早期Curci等人报道的手性酮Fig.2 It has been reported earlier Curci chiral ketone
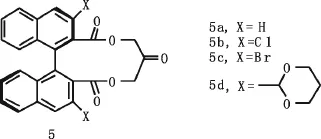
图3 杨丹等报道的手性联萘基系列酮Fig.3 Dan Yang et al reported chiral binaphthyl ketone series

图4 Denmark等报道的手性七元环酮Fig.4Denmark reported chiral seven-membered ring ketone
1996年,史一安报道的果糖衍生的手性酮9在手性酮催化环氧化的史册上写下了影响深远的篇章[9].这种酮能够从价格便宜易得的D-果糖7通过两步反应得到(见图5).
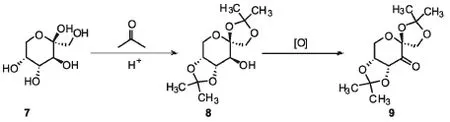
图5 经典史一安酮及其合成路线Fig.5 The classic amine ketone and its synthetic route
酮9的设计上具有这几个优点:(1)手性控制因素在发生反应的羰基旁边,增强了在底物与催化剂作用时对立体效应的控制;(2)并环和羰基邻位的季碳中心可起到有效减小立体中心消旋化的作用;(3)C2或者潜在的C2对称因素控制了烯烃底物接近Dioxirane的途径;(4)吸电子基团的引入增加了酮羰基的反应活性并降低了竞争的Baeyer-Villiger重排反应活性.
在有机不对称环氧化中,一般使用过硫酸钾复合盐——Oxone作为氧化剂.在反应液的pH值过高时,Oxone会很快的自分解10,导致反应转化率很低.而在pH<8时,又会发生催化剂酮的Baeyer-Villiger重排反应(见图6).
因为产物13和14非常容易水解,所以在前人文献中没有被分离过,但今认为,这是酮最有可能的分解途径.在稍高于pH=8条件下,阴离子中间体11形成之后立即转化为Dioxirane 12,此时12亦能通过10发生Baeyer-Villiger氧化,为了让酮9与Oxone的反应速率比其自分解的速度快,经过一系列的研究,如今的环氧化一般用缓冲溶液控制在pH=10.5的条件下反应[11].
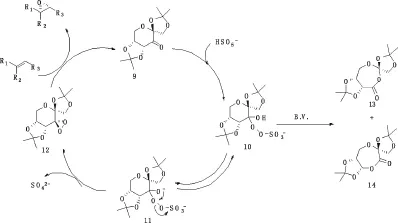
图6 酮9催化的环氧化机理Fig.6 Epoxidation catalytic mechanism of ketone 9
考虑到酮的副反应,一般酮9的用量是30mol%,对于非官能团化烯烃的不对称环氧化能获得大于90%的ee值.这种催化剂对反式1,2-二取代以及非共轭烯烃、三取代烯烃、二烯、烯炔和羟基烯烃的环氧化均能获得大于90%的ee值[12].而反式1,2-二取代及非共轭烯烃对Jacobsen-Katsuki反应是不利的[5],通常情况下高烯丙基醇或双高烯丙基醇是Sharpless环氧化不利的底物[4].典型的环氧化条件为:烯烃(1eq)、酮(0.3eq)、Oxone(1.5eq)和K2CO3(3~4eq)在乙腈与缓冲溶液中反应.具有代表性的烯烃不对称环氧化例子见图7.
酮9最好的一个应用是Corey和Xiong从双羟基角鲨烯15a仅仅通过两步双羟基化15a合成五环[15]oxasqualenoid glabrescol 15c(见图8).在这个合成过程中,五烯15a对映选择性地转化成五环氧化物15b是关键步骤,在樟脑磺酸作用下,后者重排成五环目标分子15c.
由于经典的史一安酮9只能有效地催化环氧化反式烯烃和三取代烯烃,所以史一安等人继续研究果糖衍生的氨基甲酸酯酮16(见图9),这种酮能够有效的催化顺式烯烃以及末端烯烃取得较好的ee,同时大大减少了催化剂的用量[16](16a能使催化剂用量降至1~5mol%).
2 手性亚胺盐催化不对称环氧化
手性亚胺盐催化不对称环氧化是通过手性氮杂环氧离子中间体进行反应的反应,如图10所示.
在过硫酸氢钾复合盐(Oxone)等氧化剂作用下,手性亚胺正离子原位产生不稳定的寿星氮杂氧离子中间体,手性氮杂氧离子中间体选择氧原子,并转移到碳碳双键的一个面上,同时手性亚胺正离子再生,催化循环得以进行.由于手性亚胺正离子不会像手性酮一样发生Baeyer-Villiger反应而氧化分解,催化剂用量理论上会比酮少很多,所以手性亚胺正离子的催化环氧化引起化学家的关注.
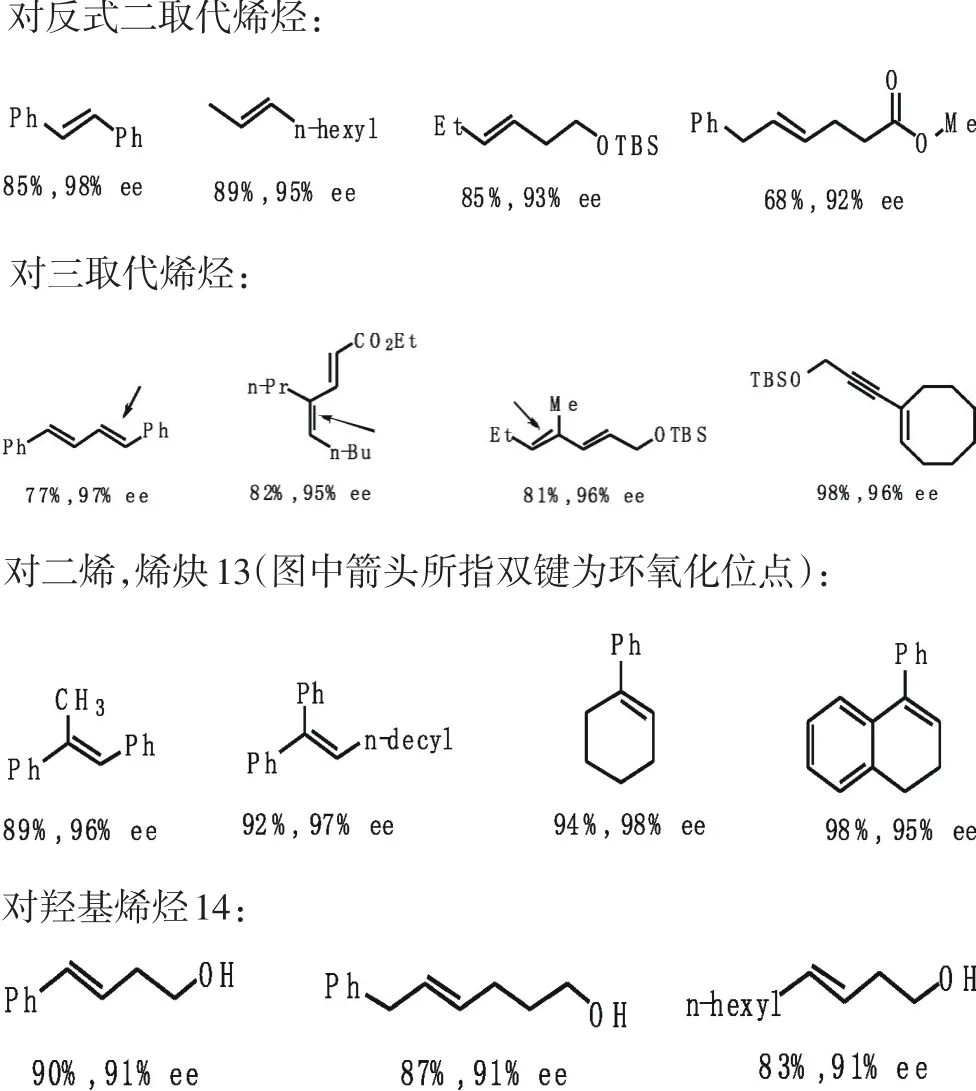
图7 具有代表性的烯烃不对称环氧化例子Fig.7The typical olefins asymmetric epoxidation example
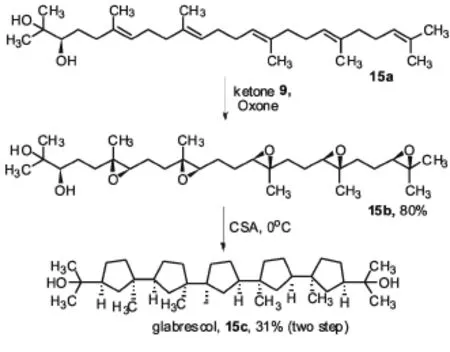
图8 oxasqualenoid glabrescol的合成Fig.8 The synthesis of oxasqualenoid glabrescol
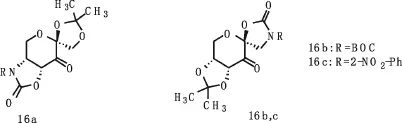
图9 氨基甲酸酯酮Fig.9Carbamate
Lusinchi等人[17]在1976和1981年报道了甾族氮杂氧离子盐19(见图11).1987年,Hanquet等人[18]制备了另一例氮杂氧离子盐20(见图11),并且在1988年发现了原位产生的催化剂20能够催化烯烃的环氧化[19],之后,手性氮杂氧离子盐催化环氧化得到了发展.
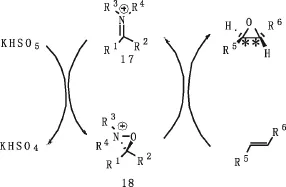
图10 手性亚胺盐催化不对称环氧化机理Fig.10 Chiral imine salt catalytic asymmetric epoxidation mechanism
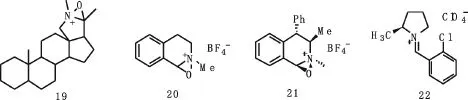
图11 最早期报道的氮杂氧离子盐Fig.11 The early reports of nitrogen impurity oxygen ionic salt
1993年,Bohe等人分离出光学纯的氮杂氧离子盐21(见图11),并用其催化一些烯烃的不对称环氧化[20],如反式二苯乙烯的不对称环氧化.纯21催化时,可以取得63%的产率和42%的ee值,用原位产生的21催化时,可以取得80~90%的转化率和35%的ee值.
1997年,Armstrong等报道了环外亚胺阳离子22催化的不对称环氧化[21],但由于环外亚胺正离子的双键极易水解,催化剂用量需要100 mol%,且ee值也只达到22%.
1998年,Page等人[22]利用普通的潜手性溴代醛前体以及易得的手性一级胺制备了一系列活性亚胺盐23(见图12).
对于大多数烯烃底物,催化剂亚胺盐23的用量只需要0.5~10 mol%,环氧化产生的ee值在30~60%之间.而反式二苯乙烯在催化剂23a的作用下以78%的产率和73%的ee值得到环氧化产物.
传统的亚胺盐催化环氧化都是用Oxone作为氧化剂,Oxone的溶解需要水.一般在低温下可以取得较好的ee值,而溶剂在低于-8℃时会结冰,导致这一类催化反应无法在更低的温度下进行.为了解决这个问题,2004年,Page等人引入了一种可以很好地溶解在有机溶剂中的无水介质氧化剂四苯基膦过硫酸盐[23]——TPPP,并发现氯仿存在下的催化反应效果比传统溶剂乙腈要好,TPPP及其合成方法见图13.
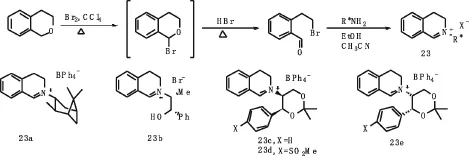
图12 亚胺盐22及其制备路线Fig.12 Imide salt Route 22 and its preparation

图13 无水介质氧化剂TPPP的合成Fig.13Synthesis of nonanhydrous condition oxidant TPPP
在1996年Aggrwal小组[24]和2004年Page小组[25]分别制备联萘基衍生的亚胺盐24a和24b(见图14),亚胺盐催化烯烃的环氧化数据中,一般的ee值为16~84%,当使用1-苯基-3,4-二氢萘作为底物,24b作为催化剂时,最高的对映选择性ee值达到95%,大大缩短了反应时间.
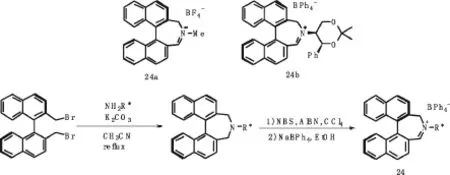
图14 联萘基衍生的亚胺盐及其合成路线Fig.14 Binaphthyl salts derived imine routes for their synthesis
迄今为止,Page等报道25a是反应速率最快的亚胺盐催化剂.一般催化氧化只需要1~10 min,ee值为40~67%.Lacour[26-27]等人将25a的抗衡阴离子换成TRISPHAT的亚胺盐25b(见图15),亲油基团TRISPHAT能够让亚胺盐留在有机相中,催化剂在一定程度上提高了对映选择性,对一系列底物取得了50~67%的ee值.
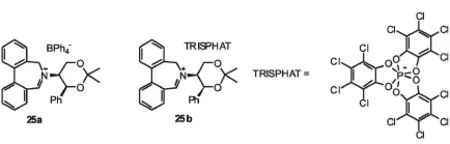
图15 联苯衍生的亚胺盐Fig.15 Imide salt derived from biphenyl
3 结论
在本文中,总结了两类碳碳双键的不对称环氧化方法.首先是以手性酮与过硫酸氢钾等氧化物原位生成二氧杂环丙烷实现的不对称环氧化.在这一领域许多华人科学家作出了突出的贡献,比如杨丹院士第一个用该方法完成了高ee值的环氧化,史一安老师使用果糖衍生物实现了便宜易得的不对称环氧化,并且该方法在全合成中得到了广泛的应用.与二氧杂环丙烷不对称氧化同时发展的是手性氮杂氧离子中间体介导的双键不对称环氧化,虽然该方法可避免二氧杂环丙烷不对称环氧化中的拜尔-威利格副反应,但该方法还未能达到广泛的底物实用性及稳定的高ee值,所以还具有很大的发展潜力.
[1]Curci R,Fiorentino M,Troisi L,et al.Epoxidation of alkenes by dioxirane intermediates generated in the reaction of potassium caroate with ketones[J].Org Chem,1980,45(23):4758-4760.
[2]Wong A O,Shi Y.Organocatalytic Oxidation.Asymmetric Epoxidation of Olefins Catalyzed by Chiral Ketones and Iminium Salts[J].Chem Rev,2008,108(9):3958-3987.
[3]Curci R,Fiorentino M,Serio M R.Asymmetric epoxidation of unfunctionalized alkenes by dioxirane intermediates generated from potassium peroxomonosulphate and chira ketones[J].J Chem Soc Chem Commun,1984:155.
[4]Curci R,D’Accolti,Fiorentino L,et al.Enantioselective epoxidation of unfunctionalized alkenes using dioxiranes generated in situ[J].Tetrahedron Lett,1995,36(32):5831-5834.
[5]Yang D,Yip Y C,Tang M W,et al.Symmetric Chiral Ketone for Catalytic Asymmetric Epoxidation of Unfunctionalized Olefins[J].J Am Chem Soc,1996,118(2):491-492.
[6]Yang D,Wong M K,Yip Y C,et al.Design and Synthesis of Chiral Ketones for Catalytic Asymmetric Epoxidation of Unfunctionalized Olefins[J].J Am Chem Soc,1998,120(24):5943-5952.
[7]Denmark S E,Wu Z.Indium(III)Chloride Promoted Highly Efficient Tandem Rearrangement-α-Addition Strategy towards the Synthesis of α-Hydroxyamides[J].Synlett,1999:847.
[8]Denmark S E,Matsuhashi H J.Chiral Fluoro Ketones for Catalytic Asymmetric Epoxidation of Alkenes with Oxone[J].Org Chem,2002,67(10):3479-3486.
[9]Tu Y,Wang Z X,Shi Y.An Efficient Asymmetric Epoxidation Method for trans-Olefins Mediated by a Fructose-Derived Ketone[J].J Am Chem Soc,1996,118(40):9806-9870.
[10]Montgomery R E.Catalysis of peroxymonosulfate reactions by ketones[J].J Am Chem Soc,1974,96(25):7820-7821.
[11]Shu L,Shi Y.An Efficient Ketone-Catalyzed Epoxidation Using Hydrogen Peroxide as Oxidant[J].J Org Chem,2000,65(25):8807-8810.
[12]Wang Z X,Tu Y,Frohn M,et al.An Efficient Catalytic Asymmetric Epoxidation Method[J].J Am Chem Soc,1997,119:11224-11235.
[13]Warren J D,Shi Y.Chiral Ketone-Catalyzed Asymmetric Epoxidation of 2,2-Disubstituted Vinylsilanes[J].J Org Chem,1999,64:7675-7677.
[14]Wang Z X,Shi Y.A pH Study on the Chiral Ketone Catalyzed Asymmetric Epoxidation of Hydroxyalkenes[J].J Org Chem,1998,63(9):3099-3104.
[15]Corey E J,Xiong Z.Simple Enantioselective Total Synthesis of Glabrescol,a Chiral C2-Symmetric Pentacyclic Oxasqualenoid[J].J Am Chem Soc,2000,122(38):9328.
[16]Tian H,She X,Shu L,et al.Highly Enantioselective Epoxidation of cis-Olefins by Chiral Dioxirane[J].J Am Chem Soc,2000,122(46):11551-11552.
[17]Milliet P,Picot A,Lusinchi X.Formation d'un sel d'oxaziridinium quaternaire par methylation d'un oxazirannemise en evidence de ses proprieties oxydantes[J].Tetrahedron Lett,1976,17(19):1573-1576.
[18]Hanquet G,Lusinchi X,Milliet P.Peracid oxidation of an immonium fluoroborate a new example of oxaziridinium salt[J].Tetrahedron Lett,1987,28(48):6061-6064.
[19]Hanquet G,Lusinchi X,Milliet P.Transfert d'oxygene sur la double liaison ethylenique a partir d'un sel d'oxaziridinium[J].Tetrahedron Lett,1988,29(32):3941-3944.
[20]Bohe'L,Hanquet G,Lusinchi M,et al.The stereospecific synthesis of a new chiral oxaziridinium salt[J].Tetrahedron Lett,1993,34(45):7271-7274.
[21]Armstrong A,Ahmed G,Garnett I,et al.Highly stereoselective intramolecular epoxidation in unsaturated oxaziridines[J].Synlett,1997:1075.
[22]Page P C B,Rassias G A,Bethell D,et al.New System for Catalytic Asymmetric Epoxidation Using Iminium Salt Catalysts[J].J Org Chem,1998,63(8):2774-2777.
[23]Page P C B,Barros D,Buckley B R,et al.Organocatalysis of Asymmetric Epoxidation Mediated by Iminium Salts under Nonaqueous Conditions[J].J Org Chem,2004,69(10):3595-3597.
[24]Aggarwal V K,Wang M F.Catalytic asymmetric synthesis of epoxides mediated by chiral iminium salts[J].Chem Commun,1996:191-192.
[25]Page P C B,Buckley B R,Blacker A.Iminium Salt Catalysts for Asymmetric Epoxidation:The First High Enantioselectivities[J].J Org Lett,2004,6(10):1543-1546.
[26]Page P C B,Buckley B R,Blacker A.Iminium Salt Catalysts for Asymmetric Epoxidation:The First High Enantioselectivities[J].J.Org Lett,2006,8(20):4669.
[27]Goncalves M H,Martinez A,Grass S,et al.Enantioselective olefin epoxidation using homologous amine and iminium catalysts—a direct comparison Lacour[J].J Tetrahedron Lett,2006,47(30):5297-5301.
责任编辑:刘 红
Catalytic Asymmetric Epoxidation of Organic Molecules
MA Long1,CHEN Shou2,ZHOU Wenli1,ZHANG Yanchen1,JU Xuecheng2,MENG Xiang1,PENG Qinqin1,ZHENG Chao1,2*
(1.School of Chemistry and Chemical Engineering,Hainan Normal University,Haikou 571158,China;2.Shenzhen Science and Technology Innovation Center,Shenzhen 518110,China)
Optically active epoxy compound is an important intermediate for organic synthesis.Via asymmetric epoxidation reaction,prochiral olefin can convert into epoxy compound which having a chiral carbon.This reaction has important significance in medicine,pesticides,and synthetic fragrances.Small organic molecule catalyzed asymmetric epoxidation of olefins is an efficient and green synthesis of optically active epoxy compound,now the reaction is mainly use chiral ketones and chiral imide salt as effective catalyst.This article describes the catalytic mechanism of such reactions,and summarizesthe research progress in these two types of asymmetric epoxidation catalytic.
asymmetric epoxidation;chiral ketones;chiral iminium
O 621.3+4
A
1674-4942(2016)02-0179-05
2016-03-02
2015年海南省自然科学基金(20152038)
*通讯作者
猜你喜欢
分子催化(2022年1期)2022-11-02
中国生殖健康(2020年5期)2021-01-18
中国生殖健康(2018年5期)2018-11-06
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
中国民族医药杂志(2016年5期)2016-05-09
中国塑料(2015年6期)2015-11-13
中国当代医药(2015年7期)2015-03-01
分析化学(2014年7期)2014-12-13
郑州大学学报(理学版)(2014年3期)2014-03-01
