
钴(III)多吡啶配合物与DNA对接性质的理论研究
2016-05-12 00:55:08苗青青
化学研究 2016年2期
苗青青
(池州学院 化学与材料工程学院,安徽 池州 247000)
钴(III)多吡啶配合物与DNA对接性质的理论研究
苗青青*
(池州学院 化学与材料工程学院,安徽 池州 247000)
摘要:采用密度泛函理论,在真空和溶液中对两个钴多吡啶配合物的几何结构进行了优化,结果表明在溶液中优化的几何结构和实验结果吻合较好. 以在溶液中优化的几何结构为基础,运用对接软件Dock 6.0,把这两个钴(III)多吡啶配合物对接到DNA碱基对中,从总体模型上,研究钴(III)多吡啶配合物与DNA碱基对作用的部位,并详细解释了配合物1与DNA键合常数大于配合物2的原因.
关键词:钴多吡啶配合物;电子结构;对接模型
钴多吡啶配合物对DNA识别及其相互作用性质的研究一直受到国内外科学家的关注[1-4],因为有些钴多吡啶配合物与DNA有较强的结合能力,这些配合物可以用来探索DNA的结构[5];有些钴多吡啶配合物在光的照射下能强烈地裂解DNA,可以用来开发高效DNA光裂解试剂[6];有些钴多吡啶配合物在水溶液中不发光,而在DNA存在下发出极强的荧光,可以用做DNA分子“光开关”[7].
为了探索钴多吡啶配合物与DNA相互作用性质,近年来大量的钴多吡啶配合物被合成,其DNA作用性质被表征. 实验结果表明钴多吡啶配合物与DNA结合有三种作用方式[5],即静电结合、沟面结合和插入结合,其中插入结合研究者最多,这种结合方式钴多吡啶配合物的主配体插入到DNA碱基对中,钴多吡啶配合物与DNA具有较强的键合力,具有这种作用方式的钴多吡啶配合物往往有较好的DNA相互作用性质,尽管实验上对这种作用方式进行大量的研究,其作用细节还不清晰,从理论上探索钴多吡啶配合物与DNA作用方式及作用细节,还是一项很有意义的工作,这些研究结果将为设计和合成高效、高选择性且靶向DNA的钴多吡啶配合物提供理论参考.
1计算方法
要研究的钴多吡啶配合物[8][Co(phen)2HPIP]3+(1)和[Co(phen)2HNAIP]3+(2)的结构示意图见图1 (phen=1,10phenanthroline, HPIP=2(2-hydroxyphenyl)imidazo[4,5f][1,10]phenanthroline, HNAIP= 2(2hydroxylnaphthyl)imidazo[4,5f][1,10]phenanthroline),可以看出,这两个钴多吡啶配合物都是由辅助配体(phen)、主配体(HPIP或者HNAIP)和一个钴原子(Co)组成变形的八面体. 运用Gaussian03量子化学程序包[9],在B3LYP/LanL2DZ水平上,在真空中对这两个钴多吡啶配合物进行几何全优化,并进行了频率分析,结果不出现虚频,表明优化的几何结构是稳定结构. 另外,由于钴原子带有3个正电荷,其几何结构受溶剂效应影响较大,为了获得更准确的几何结构,以水作为溶剂,并采用连续极性导体模型(CPCM)[10],在B3LYP/LanL2DZ水平上对这两个配合物进行了优化. 为了研究配合物与DNA的相互作用,采用对接软件Dock 6.0[11],把在溶液中优化得到的两个配合物的几何结构对接到DNA碱基对中(DNA来自于蛋白质数据库,entry 454D),对接盒子边长设定为2.0 nm,空间格子步长设定为0.03 nm,能量缺省值距离为999.9 nm,最大构象搜索设定为2×106圈,其他参数如无特殊标注均采用默认值.
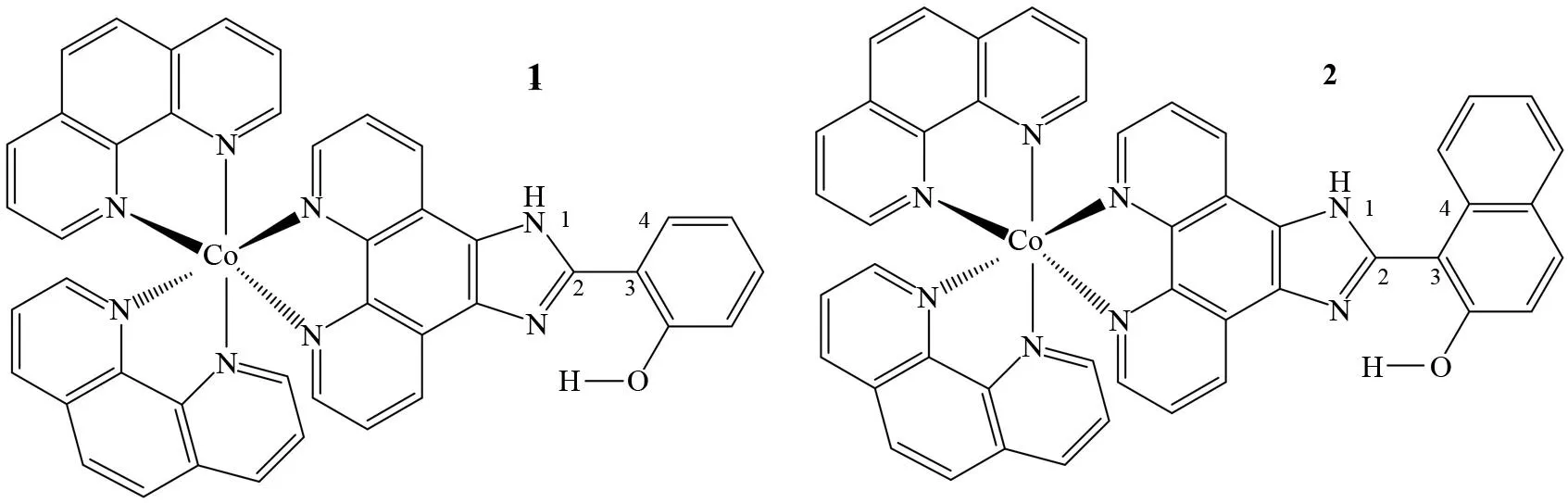
图1 配合物1和2的结构示意图和原子编号Fig.1 Structural diagrams and atomic labels of the complexes 1 and 2
2结果和讨论
2.1配合物的几何结构
用密度泛函方法在真空和溶液中分别对这两个钴多吡啶配合物进行了优化,优化的结果见表1. 由于这两个配合物都没有晶体实验结构数据,优化的几何结构不能和晶体结构数据进行比较,为了和晶体结构数据比较,以有晶体结构数据的类似配合物[12][Co(phen)3]3+为例,采用相同的计算水平分别在真空和水溶液中进行优化,计算结果也列于表1,在真空中计算得到的金属钴原子和N原子平均键长Co-N为0.198 1 nm,在水溶液中计算值为0.196 8 nm,而实验值为0.194 3 nm,说明在水溶液中的计算结果和实验结果吻合较好,同时可以看出,在真空和溶液中的计算结果差别较大,这说明溶剂效应对带有三价正电荷的钴多吡啶配合物影响较大,对于钴多吡啶配合物的几何构型优化,考虑溶剂效应是必要的.
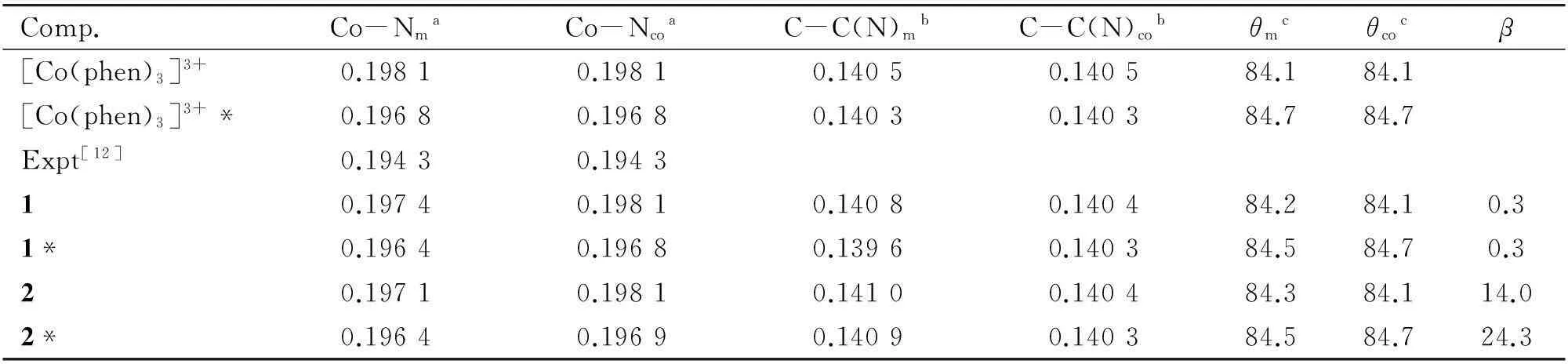
表1 在真空和溶液中计算得到的配合物1和2的键长(nm)、键角(°)和二面角(°)
aCo-Nm表示钴原子和主配体(HPIP或者HNAIP )上氮原子之间的平均键长, Co-Nco表示钴原子和辅助配体(phen)上氮原子之间的平均键长;bC-C(N)m表示主配体骨架平均键长,C-C(N)co表示辅助配体骨架平均键长;cθm表示钴原子和主配体上两氮原子之间的平均配位键角,θco表示钴原子和辅助配体上两氮原子之间的平均配位键角;β表示二面角N1-C2-C3-C4;“*”表示在溶液中优化.
对于配合物1和2,从溶液中优化的结果可以看出,钴原子和氮原子之间的平均键长、钴原子和两氮原子之间的平均键角、主配体和辅助配体上的平均键长都变化不大,在溶液中优化的几何结构,配合物1和2主配体上的二面角N1-C2-C3-C4分别为0.3°和24.3°,说明配合物1主配体的平面性好于配合物2主配体的平面性.
2.2配合物与DNA的对接性质
采用对接软件Dock6.0,把配合物1和2在溶液中优化得到的几何结构对接到DNA碱基对中,对接结果见图2. 从图2可以看出,配合物1和2的主配体都从DNA的大沟插入到DNA的碱基对中,这与实验上测得的这两个配合物都以插入的方式与DNA结合是一致的[8].
配合物1和2的平均能量得分值分别为-75.35和-74.87 kcal·mol-1,表明配合物1与DNA结合后稳定性好于配合物2,即配合物1与DNA结合能力强于配合物2,这与实验上测得配合物1和2与DNA的键合常数相一致,即Kb(1, 4.1×105L/mol) >Kb(2, 1.8×105L/mol)[8].
配合物1与DNA键合力强于配合物2的主要原因可能是配合物2的主配体平面性较差,造成配合物2的主配体插入到DNA碱基对之间时受到的阻力较大,从而导致配合物2的主配体插入到DNA碱基对之间的深度不够,因此配合物1与DNA结合力强于配合物2.
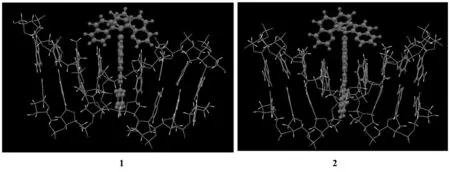
图2 配合物1和2的DNA对接模型Fig.2 DNA-docking models of complexes 1 and 2
3结论
对两个钴(III)多吡啶配合物[Co(phen)2HPIP]3+和[Co(phen)2HNAIP]3+进行了理论研究. 首先,对这两个钴(III) 多吡啶配合物在真空和溶液中进行优化,在水溶液中优化得到的几何结构和实验值吻合较好,而在真空中优化得到的几何结构和实验值偏离较大,说明水溶剂效应对这类钴多吡啶配合物的几何结构影响较大. 其次,用对接软件Dock6.0,把配合物1和2在溶液中优化得到的几何结构对接到DNA碱基对中,结果表明,配合物1和2的主配体都能插入到DNA碱基对之间,说明配合物1和2都以插入的方式与DNA结合,与实验结果吻合较好. 最后,根据对接模型,解释了配合物1和2与DNA键合强或者弱的原因.
参考文献:
[1] ZHANG Q L, LIU J G, CHAO H, et al. DNA-binding and photocleavage studies of cobalt(III) polypyridyl complexes: [Co(phen)2IP]3+and [Co(phen)2PIP]3+[J]. J Inorg Biochem, 2001, 83: 49-55.
[2] MIAO T F, LI F, LI J, et al. Theoretical studies on the related properties of Co(III) polypyridyl complexes interacting with DNA [J]. J Inorg Biochem, 2012, 109: 16-25.
[3] MIAO T F, LI F, XU L C, et al. Theoretical studies on the DNAintercalator properties of Co(III) polypyridyl complexes [J]. Comput Theor Chem, 2011, 976: 209-214.
[4] MIAO T F, LI J, LIAO S Y, et al. Theoretical studies on DNA binding, DNA photocleavage and spectral properties of Co(III) complexes [Co(phen)2(L)]3+(L=pip, hpip, hnaip) [J]. Inorg Chim Acta, 2010, 363: 3880-3886.
[5] JI L N, ZOU X H, LIU J G. Shape- and enantioselective interaction of Ru(II)/Co(III) polypyridyl complexes with DNA [J]. Coord Chem Rev, 2001, 216/217: 513-536.
[6] SASTRI C V, ESWARAMOORTHYL D, GIRIBABU L, et al. DNA interactions of new mixed-ligand complexes of cobalt(III) and nickel(II) that incorporate modified phenanthroline ligands [J]. J Inorg Biochem, 2003, 94: 138-145.
[7] MOUCHERON C, MESMAEKER A K, CHOUA S. Photophysics of Ru(phen)2(PHEHAT)2+: A novel “Light Switch” for DNA and photo-oxidant for mononucleotides [J]. Inorg Chem, 1997, 36: 584 -592.
[8] ZHANG Q L, LIU J G, LIU J Z, et al. Effect of intramolecular hydrogen-bond on the DNA-binding and photocleavage properties of polypyridyl cobalt(III) complexes [J]. Inorg Chim Acta, 2002, 339: 34-40.
[9] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 03[CP]. Revision D.01, Wallingford CT: G aussian, Inc., 2005.
[10] BARONE V, COSSI M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model [J]. J Phys Chem A, 1998, 102: 1995-2001.
[11] SHOICHET B K, KUNTZ I D, BODIAN D L. Molecular docking using shape descriptors [J]. J Comput Chem, 1992, 13: 380-397.
[12] NIEDERHOFFER E C, MARTELL A E, RUDOLF P, et al. Tris(1,10-phenanthroline) cobalt(III) triperchlorate dihydrate, [Co(C12H8N2)3]·2H2O [J]. Cryst Struct Communic, 1982, 11: 1951-1957.
[责任编辑:吴文鹏]
Theoretical study on docking properties of cobalt (III) polypyridyl complexes with DNA
MIAO Qingqing*
(CollegeofMaterialsandChemicalEngineering,ChizhouUniversity,Chizhou247000,Anhui,China)
Abstract:Full geometry optimizations of two polypyridyl complexes have been carried out in vacuum and in aqueous solution using the density functional theory (DFT). The results show that the optimized geometric structures in aqueous solution are in considerable agreement with the experimental results. On the basis of the DFT optimized ground geometry in aqueous solution, the two Co(III) polypyridyl complexes were docked with DNA base pairs using docking software Dock 6.0. The interaction sites on DNA of these complexes were deeply studied based on the whole DNA-complex model and the trend in DNA-binding affinities, i.e., Kb(1) > Kb(2), was reasonably explained.
Keywords:Co(Ш) polypyridyl complex; electronic structure; docking model
文章编号:1008-1011(2016)02-0161-04
中图分类号:O641.121
文献标志码:A
作者简介:苗青青(1995-),女,本科生,主要从事生物无机理论化学研究. *通讯联系人,E-mail: qingqmiao@126.com.
收稿日期:2016-02-19.
