
细胞焦亡在心力衰竭进程中的研究进展*
2016-05-07 02:33胡厚祥陈海燕
西部医学 2016年3期
胡厚祥 陈海燕
细胞焦亡在心力衰竭进程中的研究进展*
胡厚祥 陈海燕
(川北医学院附属医院心内科 , 四川 南充 637000)
细胞焦亡(pyroptosis)是一种新的程序性细胞死亡方式,主要通过半胱天冬氨酸蛋白酶-1(caspase-1)来介导,并伴有大量的促炎性细胞因子的释放。在心力衰竭进展的不同阶段,细胞焦亡通过炎症小体活化及激活caspase-1参与心力衰竭的病理生理过程。本文就细胞焦亡在心力衰竭进程中的作用作一评述。
细胞焦亡; 心力衰竭; NLRP3 炎症小体; caspase-1
执行编委简介:胡厚祥,主任医师,医学博士,硕士生导师,加拿大留学归国人员,川北医学院附属医院心内科副主任,川北医学院学术和技术带头人,四川省第十一批有突出贡献优秀专家,四川省医学会心血管分会委员,四川省医学会临床流行病学分会常委。担任国家自然科学基金一审评审专家,《西部医学》编委,《American Journal of Hypertension》及《PLOS ONE》杂志的审稿专家。先后主持国家自然科学基金面上项目、教育部留学基金委、四川省科技厅、四川省教育厅和四川省卫生厅科研项目10余项。发表论文60余篇,其中SCI论文7篇。近年主持研究的科研成果项目获四川省科技进步一等奖、四川省医学会科技进步一等奖和南充市科技进步一等奖各1项,四川省科技进步三等奖1项。
心力衰竭(heart failure,HF,心衰)是各种心血管疾病的最终归宿,根据其不同临床进展分为前心衰阶段(有心衰的高危因素,心脏无结构异常)、前临床心衰阶段(有结构性心脏病,但没有心衰的临床症状体征)、临床心衰阶段(有结构性心脏病,并有心衰的临床症状和体征)和难治性终末期心衰阶段。从冠状动脉粥样硬化症到冠状动脉粥样硬化性心脏病(冠心病)、再进展到心肌梗死、心室重构和心衰的病理生理过程中,心肌细胞、血管平滑肌细胞和内皮细胞在高胆固醇、缺血缺氧等作用下发生一系列应激反应导致的细胞死亡起着非常重要的作用。细胞焦亡是最近定义的一种程序性细胞死亡形式,其发生伴随着大量促炎性危险相关分子模式(DAMPs)的释放,引起邻近细胞的炎症反应和粒细胞浸润,进而导致和加重组织炎症反应。近年来,细胞焦亡在心衰进程中的作用受到大家关注。本文就细胞焦亡在心力衰竭进程中的作用作一评述,以供基础与临床研究参阅。
1 细胞焦亡
细胞焦亡的发生依赖于半胱天冬氨酸蛋白酶-1(caspase-1,在鼠类细胞中为caspase-1和caspase-11)的激活,而区别于细胞凋亡时caspase-3的活化依赖[1]。细胞焦亡兼有细胞凋亡和细胞坏死的特点,表现为核固缩、DNA片段化、细胞膜孔形成、细胞水肿、胞膜破裂和细胞内容物释放到胞外引起的炎症反应[2]。
细胞焦亡的发生是细胞内炎症小体活化的结果,包括典型性炎症小体活化和非典型性炎症小体活化两种途径。典型性炎症小体活化模型中,病原体或DAMPs通过活化NLRP3(NOD-,LRR and pyrin domain[PYD]-containing protein3, NLRP3)、NLRC4、NLRP1等炎症小体,引起caspase-1(或caspase-11)前体剪切为活性caspase-1;活化的caspase-1进一步剪切IL-1β和IL-18的前体为其成熟体,同时引起细胞的焦亡[3]。非典型炎症小体途径是指病原体或DAMPs诱导非典型性炎症小体活化,激活caspase-11,活化的caspase-11直接引起细胞焦亡或通过激活NLRP3炎症小体,进而由caspase-1介导细胞的焦亡反应。目前在介导细胞焦亡的炎症小体活化中,对NLRP3炎症小体活化的研究最为广泛和全面,其由NLRP3、ASC(Apoptosis-associated speck-like protein containing CARD, ASC)和pro-caspase-1三种蛋白组成[4]。NLRP3由C端亮氨酸富集结构域(LRRs)、中央区的核苷酸结合寡聚化域(NACHT域)和N端效应结构域(PYD)三部分组成,在病原体或DAMPs的刺激作用下,NLRP3通过PYD连接ASC,ASC通过其CARD结构域与caspase-1的CARD域结合,形成NLRP3炎症小体,最终导致caspase-1的剪切活化[5]。
细胞焦亡是一种特殊的非凋亡的程序性细胞死亡,其过程主要由NLRP3等炎症小体活化及其下游效应因子(caspase-1)的激活介导。故可通过检测NLRP3炎症小体活化及下游效应炎性因子水平、TUNEL染色、胞核的变化及DNA的连续性等来间接反应细胞焦亡。
2 细胞焦亡在心力衰竭进程中的作用
心衰的主要发病机制之一为心肌病理性重构,而导致心衰进展的有两个关键过程:一是心肌死亡(如凋亡、坏死、自噬等)的发生;二是神经内分泌系统过度激活所致的系统反应。此外,心衰伴随着轻度慢性的炎症反应,使其发展为不良的心脏重构[6]。细胞焦亡是一种促炎性的细胞死亡,这种积炎症反应和细胞死亡为一身的新型细胞死亡方式在心衰进程中也发挥着重要作用。
2.1 细胞焦亡与动脉粥样硬化症 在动脉粥样硬化斑块中,特别是晚期斑块中,可见增多的各种细胞死亡,包括巨噬细胞、血管平滑肌细胞和内皮细胞等。传统上,人们使用凋亡和坏死这一范式界定细胞死亡,但除了凋亡和坏死,还存在胀亡、焦亡等多种形式的细胞死亡。在动脉粥样硬化斑块中,很多研究已致力于建立细胞死亡与炎症反应的联系。而在电子显微镜下显示,动脉粥样斑块中大多数的死亡细胞伴有细胞裂解,并不是细胞凋亡。并且caspase-3激活在动脉粥样斑块中仅轻微升高,而敲除caspase-3或p53虽然可以抑制斑块中巨噬细胞凋亡,却会增多巨噬细胞,加重斑块的炎症反应和斑块的进一步恶化[7]。因此,一个合理的假设就是其他形式的细胞死亡(包括细胞焦亡)参与动脉粥样斑块的形成与发展。
细胞焦亡是一种新发现的caspase-1活化依赖的细胞死亡。Caspase-1的活化及其导致的细胞焦亡已被证实在免疫系统和中枢神经疾病中具有重要作用。现在越来越多的证据表明,细胞焦亡在动脉粥样硬化中,特别是进展性粥样斑块中同样发挥重要作用[8~10]。细胞焦亡最显著的特征是caspase-1活化的依赖性,而在粥样斑块中则发现caspase-1的高表达,并且与斑块进展程度相一致[11]。并且应用caspase-1抑制剂ac-YVAD-CHO能显著抑制粥样斑块的TUNEL阳性反应[7]。此外,胆固醇结晶和oxLDL可激活NLRP3炎症小体,导致caspase-1的活化,引起细胞裂解,DNA断裂,并释放IL-1β和IL-18,从而促进动脉粥样斑块的形成[12, 13]。
2.2 细胞焦亡与急性心肌梗死 炎症和细胞死亡在动脉粥样硬化中未得到控制而进一步发展将导致冠状动脉病变和急性冠脉综合症(ACS)。ACS病人中NLRP3炎症小体的水平与冠状动脉粥样硬化的程度呈正相关,可作为主要不良心血管事件的独立预测因子[14]。
急性心肌梗死除损伤区域的心肌组织发生死亡之外,还会通过炎症小体的活化产生无菌性炎症反应[15]。NLRP3炎症小体中的NLRP3蛋白通过与适配器ASC相互作用,从而募集并活化caspase-1,活化的caspase-1将IL-1β前体(Pro-IL-1β)剪切成为其活性形式IL-1β,并最终导致心肌细胞焦亡。IL-1β作为炎症反应的关键性因子,现已成为心肌梗死中早期炎症反应的突出标志。在急性心肌梗死边缘区的心肌细胞中ASC、NLRP3、caspase-1的量随时间的延长明显增多。在缺血心肌区域心肌成纤维细胞中NLRP3水平显著升高。而抑制NLRP3活化可以减少心肌梗死的面积,并保存梗死后心肌的功能[16];Txnip还通过调节心脏微血管内皮细胞中的NLRP3炎症小体,阻止Txnip/NLRP3信号通道,抑制NLRP3炎症小体发挥作用[17]。冠心病心肌梗死后即进展到前临床心衰阶段(小面积心肌梗死),甚至临床心衰阶段(大面积心肌梗死)。在此过程中作为一种促炎性的细胞死亡,心肌细胞焦亡(包括心肌细胞的死亡和心肌组织中的炎症反应)参与了心肌的损伤和心室重构。心肌细胞死亡使其心肌受损,心肌功能紊乱,心肌组织中的炎症反应进一步加重心肌的损伤,促进心肌重构。所以,在心肌梗死的治疗中,针对NLRP3炎症小体活化的干预将减少心肌细胞焦亡及心肌组织的炎症反应,可能成为潜在有效的治疗手段。
2.3 细胞焦亡与心力衰竭 心肌缺血性损伤可触发强烈的炎性反应,从而促进心脏功能紊乱和心衰的发生[18, 19]。NLRP3是细胞内的Nod样受体,作为危险信号的传感器,可被细胞内感染、ATP、尿酸、胆固醇结晶及损伤组织释放的细胞碎片激活[16]。被激活的NLRP3炎症小体进一步活化caspase-1,释放IL-1β。另外,缺血性心肌坏死后释放ATP和细胞碎片,激活膜P2X7受体通道,使K+外流,从而激活NLRP3,活化的NLRP3募集ASC和Pro-caspase-1,组装成复合体NLRP3炎症小体,从而自催化激活caspase-1,caspase-1剪切Pro-IL-1β和其他促炎性细胞因子为有活性的成熟体,进一步扩大炎性反应。此外,Caspase-1是炎症反应中的重要调制器,活化的caspase-1可触发细胞焦亡,进一步损害心肌功能而引发心肌梗死后心衰。
心肌损伤后的病理性重构是心衰发生的主要机制之一,炎性反应主要是修复心脏,但是过度的炎性反应将导致不良的心室重构而引发心衰。在心肌重构和修复中,NLRP3炎症小体活化主要发生在心肌成纤维细胞中[20]。心肌缺血性损伤后主要通过ROS产生和K+外流来激活NLRP3炎症小体,引发无菌性炎性反应,炎性成纤维细胞释放趋化因子,募集巨噬细胞和中性粒细胞等到损伤的组织区域,进一步扩大炎性反应[21]。此外,在心衰的发展过程中,NLRP3炎症小体活化后最终导致IL-1β释放,IL-1β一方面可诱发心肌肌浆网中的钙离子溢出,直接影响钙离子的内环境和心肌兴奋-收缩偶联,损害心肌收缩功能;另一面可刺激诱导型一氧化氮合酶 (iNOS)合成,引起细胞死亡和组织重塑导致心衰发生(图1)。
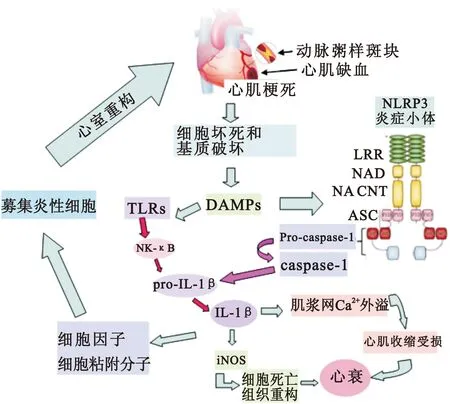
图1 急性心肌梗死激活炎性反应
Figure 1 Initiation of inflammatory response following acute myocardial infarction
3 小结与展望
在多种心血管危险因子刺激下,心血管相关细胞中炎症小体被激活,进而导致caspase-1的活化。Caspase-1的活化则进一步引起活化依赖的细胞焦亡并参与心力衰竭的发生发展。细胞焦亡是一种促炎性的细胞死亡,伴随着大量DAMPs的释放,释放的DAMPs启动炎症反应并进一步激活其它细胞的炎症小体,启动新一轮的细胞焦亡。这一循环将细胞死亡与炎症反应联系起来,加重心血管病变中的炎症反应和不稳定性。炎症小体活化和细胞焦亡也揭示在心衰发展进程的各阶段均有细胞死亡和炎症反应的参与。当然,细胞焦亡在心衰中的确切作用,以及细胞焦亡在心衰中如何发生都有待进一步的研究阐明。
[1]Duprez L, Wirawan E, Vanden Berghe T,etal. Major cell death pathways at a glance[J]. Microbes Infect, 2009, 11(13): 1050-1062.
[2]Galluzzi L, Vitale I, Abrams JM,etal. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012[J]. Cell Death Differ, 2012, 19(1): 107-120.
[3]Lim Y and Kumar S. A single cut to pyroptosis[J]. Oncotarget, 2015, 6(35): 36926-36927.
[4]Lorenz G, Darisipudi MN and Anders HJ. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis[J]. Nephrol Dial Transplant, 2014, 29(1): 41-48.
[5]Sutterwala FS, Haasken S and Cassel SL. Mechanism of NLRP3 inflammasome activation[J]. Ann N Y Acad Sci, 2014, 1319:82-95.
[6]Butts B, Gary RA, Dunbar SB,etal. The Importance of NLRP3 Inflammasome in Heart Failure[J]. J Card Fail, 2015, 21(7): 586-593.
[7]Chang W, Lin J, Dong J,etal. Pyroptosis: an inflammatory cell death implicates in atherosclerosis[J]. Med Hypotheses, 2013, 81(3): 484-486.
[8]Zheng Y, Gardner SE and Clarke MC. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease[J]. Arterioscler Thromb Vasc Biol, 2011, 31(12): 2781-2786.
[9]Yin Y, Li X, Sha X,etal. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway[J]. Arterioscler Thromb Vasc Biol, 2015, 35(4): 804-816.
[10] Son SJ, Rhee KJ, Lim J,etal. Triglyceride-induced macrophage cell death is triggered by caspase-1[J]. Biol Pharm Bull, 2013, 36(1): 108-113.
[11] Gage J, Hasu M, Thabet M,etal. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice[J]. Can J Cardiol, 2012, 28(2): 222-229.
[12] Zhang Y, Li X, Pitzer AL,etal. Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: beyond inflammation[J]. Antioxid Redox Signal, 2015, 22(13): 1084-1096.
[13] Lin J, Shou X, Mao X,etal. Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages: implication in lesion instability[J]. PLoS One, 2013, 8(4): e62148.
[14] Afrasyab A, Qu P, Zhao Y,etal. Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients[J]. Heart Vessels[J/OL], Augzo[Epub ahead of print]. PMID:26290166.2015:
[15] Fang L, Moore XL, Dart AM,etal. Systemic inflammatory response following acute myocardial infarction[J]. J Geriatr Cardiol, 2015, 12(3): 305-312.
[16] Mezzaroma E, Toldo S, Farkas D,etal. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse[J]. Proc Natl Acad Sci U S A, 2011, 108(49): 19725-19730.
[17] Liu Y, Lian K, Zhang L,etal. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury[J]. Basic Res Cardiol, 2014, 109(5): 415.
[18] Calvillo L, Vanoli E, Andreoli E,etal. Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion[J]. J Cardiovasc Pharmacol, 2011, 58(5): 500-507.
[19] Bujak M and Frangogiannis NG. The role of IL-1 in the pathogenesis of heart disease[J]. Arch Immunol Ther Exp (Warsz), 2009, 57(3): 165-176.
[20] Kawaguchi M, Takahashi M, Hata T,etal. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury[J]. Circulation, 2011, 123(6): 594-604.
[21] Chen W and Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair[J]. Biochim Biophys Acta, 2013, 1833(4): 945-953.
The role of pyroptosis in heart failure
HU Houxiang,CHEN Haiyan
(DepartmentofCardiology,NorthSichuanMedicalCollegeAffiliatedHospital,Nanchong637000,Sichuan,China)
Pyroptosis is a newly identified form of programmed cell death mediated by caspase-1. It leads to inflammatory response through the release of pro-inflammatory cytokines. Pyroptosis participates in the pathophysiological progress of heart failure through inflammasome activation and activated caspase-1.
Pyroptosis; Heart failure; NLRP3 inflammasome; Caspase-1
国家自然科学基金(81070101)
R 541.6
A
10.3969/j.issn.1672-3511.2016.03.003
2015-12-17; 修回日期: 2016-01-05; 编辑: 母存培)
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
材料与冶金学报(2022年2期)2022-08-10
现代临床医学(2022年3期)2022-06-06
医学综述(2022年7期)2022-04-19
中老年保健(2021年2期)2021-12-02
医学食疗与健康(2021年27期)2021-05-13
昆明医科大学学报(2020年12期)2021-01-26
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
