
不同OTUs划分阈值对湖泊浮游细菌α多样性的影响
2016-04-11 07:19:16王司辰曹新益赵大勇
化学与生物工程 2016年2期
沈 烽,黄 睿,王司辰,曹新益,赵大勇
(1.河海大学 水文水资源与水利工程科学国家重点实验室,江苏 南京 210098;
2.河海大学水文水资源学院,江苏 南京 210098)
不同OTUs划分阈值对湖泊浮游细菌α多样性的影响
沈烽1,2,黄睿1,2,王司辰1,2,曹新益1,2,赵大勇1,2
(1.河海大学 水文水资源与水利工程科学国家重点实验室,江苏 南京 210098;
2.河海大学水文水资源学院,江苏 南京 210098)
摘要:微生物研究中,湖泊浮游细菌微生物群落的α多样性通过OTUs(operation taxonomy units)来分析评价。考察了现有手段下不同OTUs划分阈值或分类单元“属”水平对α多样性大小和趋势的影响,并推断了分类单元“属”所对应的OTUs划分阈值。通过454高通量测序技术研究了南京莫愁湖、前湖、玄武湖、紫霞湖4个湖泊的浮游细菌微生物群落,探讨了不同OTUs划分阈值对α多样性指数OTUs、Chao1、Invsimpson、Shannon的影响。结果发现,OTUs划分阈值不同,OTUs和Chao1指数变化一致,Invsimpson和Shannon指数变化一致;在不同的OTUs划分阈值下,4个湖泊的OTUs和Chao1指数在不同季节的排序基本保持不变,Invsimpson和Shannon指数在不同季节的排序发生改变。表明,分类单元“属”所对应的OTUs划分阈值在0.06~0.08范围内;原有的以0.03或0.1阈值划分OTUs来分析α多样性是合理的;所用的分析微生物群落α多样性方法是全面的、可靠的。
关键词:α多样性;高通量序列;OTUs;浮游细菌;16S rRNA
对细菌“种”的科学且合理的定义有助于微生物学者准确认识微生物多样性时空分布特征。细菌本身的特点(如频繁的基因水平转移)使得其基因组存在着高度的动态变化,导致对细菌“种”的定义存在着巨大困难。目前关于微生物多样性的大部分研究依赖于16SrRNA基因序列的相似性来解释。通过比较序列与群落中其它未知序列间的相似性,将序列分成不同的操作性分类单元(OTUs)[2-3]。由于微生物类群之间rDNA/rRNA(细菌用16S,真菌主要用ITS)的变异速率不同,仅用16SrRNA基因序列的相似性来定义细菌的分类单元存在很大弊端[3-4],受研究方法的限制,目前常取阈值0.03或者0.1的16SrRNA基因序列差异性来划分OTUs[5]。
通过计算基于OTUs表或者种系型(种类和相对丰度)的细菌α多样性可描述细菌的多样性。其中,用于表征α多样性的指数有2大类:
1)计算菌群丰度(communityrichness)的指数
(1)OTUs指数:样品种OTUs的数量。
(2)Chao1指数:在生态学中常用来估计物种总数,是用Chao1算法估计群落中含OTUs数目的指数,由Chao于1984年最早提出。按下式计算:
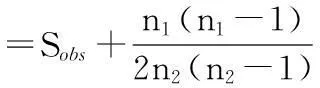
式中:Sobs为实际测量的OTUs数目;n1为只含有1条序列的OTUs数目;n2为只含有2条序列的OTUs数目。
OTUs和Chao1指数反映了微生物群落在数量上的大小程度。
2)计算菌群多样性(community diversity)的指数
(1)Invsimpson指数: Simpson指数的倒数,由Edward Hugh Simpson 于1949年提出,在生态学中常用来定量描述一个区域的生物多样性。Invsimpson指数值越大,说明群落多样性越高。按下式计算:
式中:ni为含有i条序列的OTUs数目;N为所有的序列数。
(2)Shannon指数:Shannon指数值越大,说明群落多样性越高。按下式计算:
Invsimpson指数和Shannon指数反映了微生物群落在数量结构上的分布特点。
不同的OTUs划分阈值对α多样性指数影响较大,阈值0.03或0.1的选取也存在较大的争议[6-8]。Cleary等[9]探讨了不同的OTUs划分阈值对β多样性指数的影响。但全面探讨OTUs划分阈值对α多样性的影响较少。鉴于此,作者通过454高通量测序技术研究了南京莫愁湖、前湖、玄武湖、紫霞湖4个湖泊的浮游细菌微生物群落,探讨了不同的OTUs划分阈值对α多样性指数的影响,以期用更合理的方法来阐明微生物群落的α多样性。
1实验
1.1样品采集
取南京莫愁湖、前湖、玄武湖、紫霞湖4个湖泊0.5 m深水样,从2011年11月到2012年1月在春夏秋冬4个季节各采集一次。
1.2DNA提取、PCR扩增和焦磷酸测序
用5 μm过滤器过滤水样,去除悬浮颗粒物;用0.22 μm乙酸纤维素滤网过滤滤液,收集浮游细菌细胞;按Gillan的方法[10]提取DNA。
将纯化后的基因组DNA作为PCR模板,采用通用引物533R(5′-TTACCGCGGCTGCTGGCAC-3′)和27F(5′-AGAGTTTGATCCTGGCTCAG-3′)对细菌的16S rRNA进行扩增。PCR引物上固定一个10 bp长度的barcode,用于区分不同的样品。PCR反应体系(50 μL):10 μL 5×Prime STAR buffer(plus Mg2+),0.2 mmol·L-1dNTPs,0.4 μmol·L-1上下游引物,2.0 U TaKaRa酶,加ddH2O至50 μL。PCR扩增程序:94 ℃ 5 min;94 ℃ 30 s,55 ℃ 30 s,72 ℃ 30 s,24个循环;72 ℃延伸7 min。每个样品做3个重复,然后充分混合。PCR产物用2.0%琼脂糖凝胶电泳检测,并用AxyPrep DNA试剂盒(杭州爱思进生物技术股份有限公司)纯化。
PCR扩增结果送至上海美吉公司在罗氏454FLX Titanium平台上测序。本研究所有的测序结果均被上传到美国国家生物信息中心(NCBI)数据库(http://www.ncbi.nlm.nih.gov/),检索号为SRP064272。
1.3序列处理
序列处理通过Mothur软件包完成,流程如下:序列结果通过454标准化操作(SOP)完成。下机原始高通量数据,通过降噪和修剪[11]后,删除低质量(平均质量<27)、较短(<200 bp)的不包含引物和barcode的序列,并删除单一碱基连续出现8次以上的序列[12]。剩余序列被反转并通过NAST算法与已知 SILVA 16S rRNA 基因数据库进行对齐[13]。使用Mothur中“chimera.uchime”命令去除嵌合体,“pre.cluster”被用来整理数据加速距离计算过程[14]。得到的序列与RDP 16S rRNA基因数据库进行比对分类(http://rdp.cme.msu.edu),相似度达80%以上的结果才允许被使用[15]。采用furthest-neighbor算法[16]用不同的阈值划分OTUs,在序列差异0.5%~15.0%范围内每间隔0.5%划分OTUs。统一数量差异水平并获得同一比对标准,在计算α多样性指数前先对每一个样品使用“subsample”命令进行随机取样,保证每个样品具有相同的测序深度。
1.4统计分析
α多样性指数(OTUs、Chao1、Invsimpson、Shannon)通过Mothur中“summary.single”命令计算得到。微生物属热图通过R语言中“ggplot2”包计算得到。
2结果与讨论
2.1属水平上微生物α多样性分析
将得到的序列和RDP 16S rRNA基因数据库比对,得到了每一条序列所对应的分类学属(genus)的水平。由于部分序列并不能通过比对得到所对应的属水平,在分析时并未被考虑。每个样品中属水平已分类的序列占总序列的百分比见表1。通过统计每一个属在每个样品所占比例绘制的微生物属热图见图1。
表1属水平已分类的序列占总序列的百分比/%
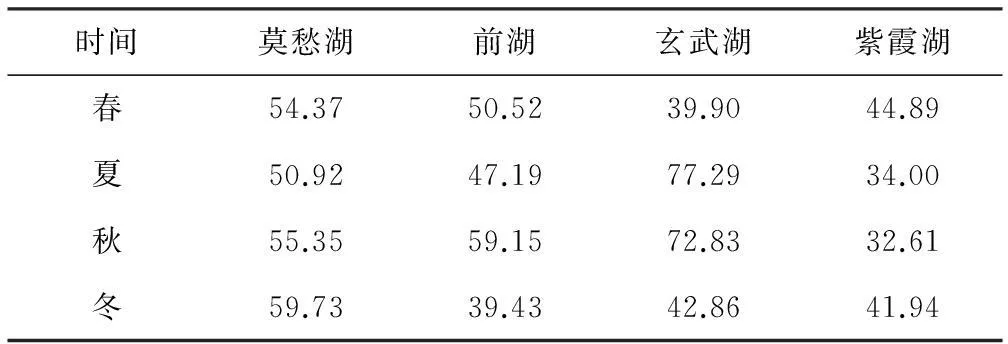
Tab.1 Relative percent of classified sequences

注:每个样品中排名前八的属被表示出来。颜色深浅表示每个属在一个样品中的相对百分比。
由图1可看出,4个湖泊在属水平上,蓝藻门细菌占主要。高比例的蓝藻直接决定了4个湖泊微生物多样性的高低。春、冬季,低比例的蓝藻使得微生物多样性较高;夏、秋季,高比例的蓝藻使得微生物多样性较低。
属水平上不同湖泊在不同季节的微生物α多样性指数(Invsimpson、Shannon)见表2。
表2属水平上的微生物α多样性指数
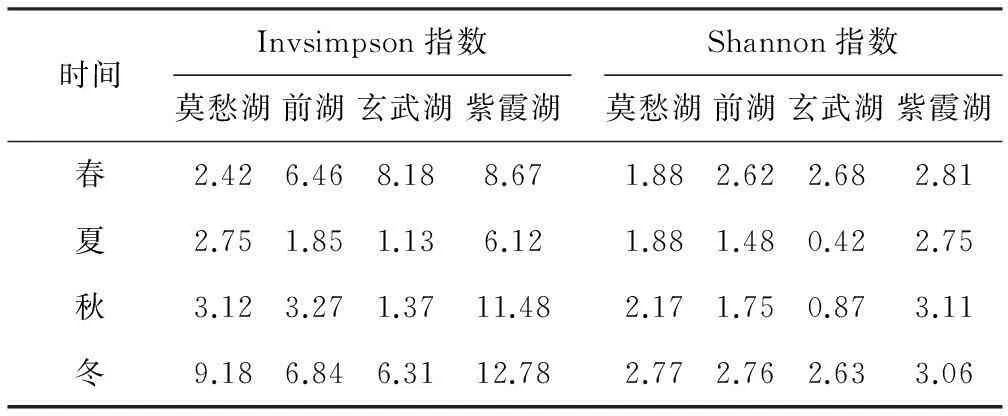
Tab.2 α-Diversity indexes of microorganisms on
由表2可看出,属水平上Invsimpson指数与Shannon指数是高度一致的,Spearman相关系数R=0.947(P<0.001),说明这2个α多样性指数在属水平上的评价结果基本一致。可以大致得到属水平上4个湖泊在不同季节的微生物α多样性表现为:莫愁湖:冬>夏≈秋>春;玄武湖:春≈冬>秋≈夏;前湖:冬>春>秋>夏;紫霞湖:冬≈秋>春>夏。
2.2不同OTUs划分阈值α多样性分析
在0.5%~15.0%范围内,每间隔0.5%的步长划分OTUs,每个样品得到了30张OTUs表。然后计算每一张表中每一个样品的α多样性指数。不同OTUs划分阈值所对应的α多样性指数(OTUs、Chao1、Invsimpson、Shannon)见图2。
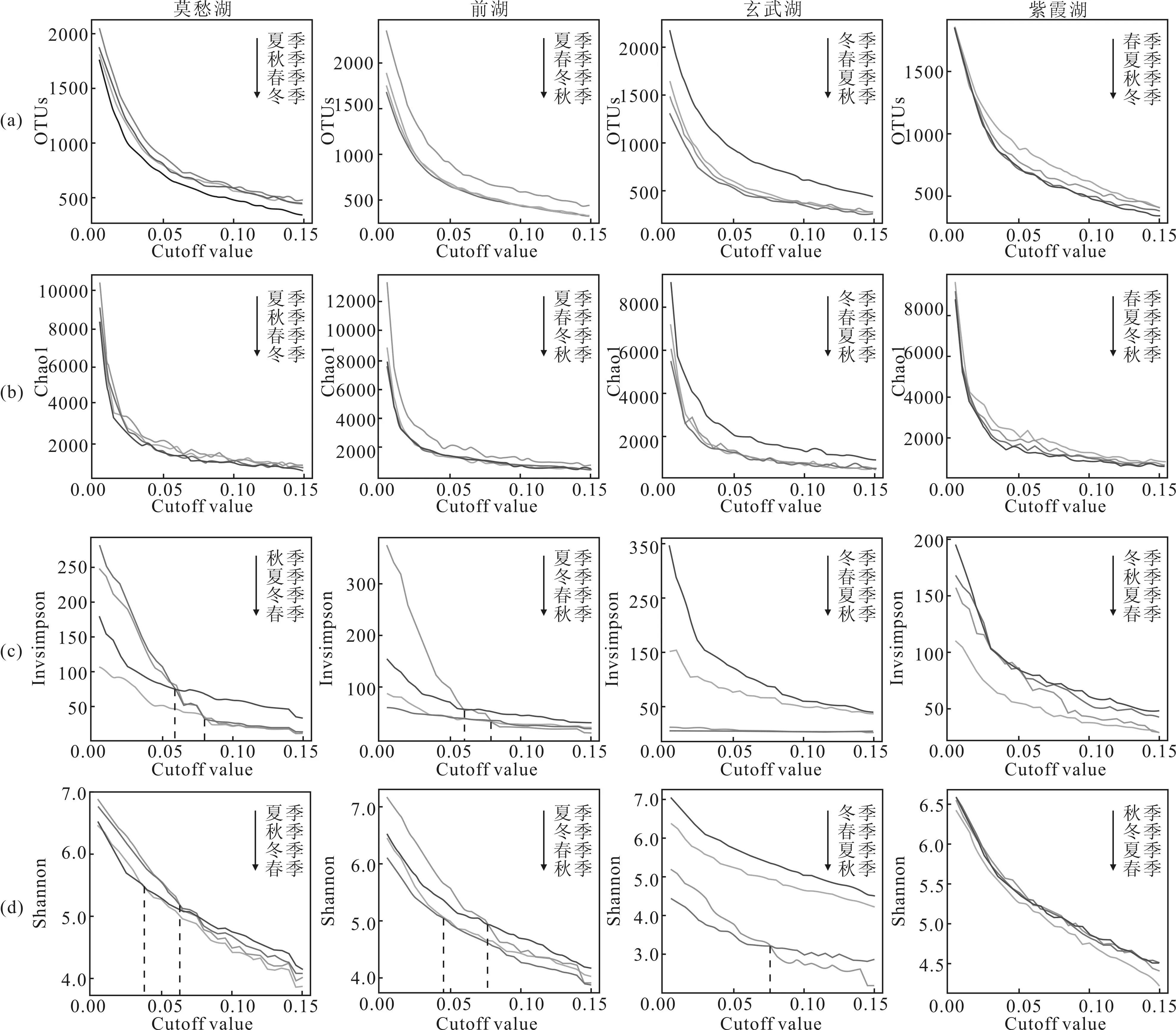
图2 不同OTUs划分阈值所对应的OTUs指数(a)、Chao1指数(b)、Invsimpson指数(c)、Shannon指数(d)
由图2a可知,随划分阈值的递进,4个湖泊在不同季节的OTUs指数逐渐降低,且递减趋势保持一致,曲线基本没有出现交叉的情况,即OTUs指数在不同季节的差异并未随划分阈值的不同而发生明显改变,说明,无论选取何种阈值来划分OTUs,4个湖泊的OTUs指数在不同季节的排序基本一致,即:莫愁湖:夏>秋≈春>冬;前湖:夏>春>冬>秋;玄武湖:冬>春>夏>秋;紫霞湖:春>夏>秋≈冬。
图2b与图2a的结果基本一致,但图2b中线条之间的交叉明显多于图2a,在区分物种α多样性时存在明显的不足。在Chao1指数差异较小时,如莫愁湖的四季、前湖的春秋冬季、玄武湖的春秋冬季,曲线交叉重叠,无法准确判断。
图2c、2d与图2a、2b有明显差异,线条出现交叉。在Invsimpson指数中,莫愁湖在划分阈值约为0.06时夏、秋线与冬季线交叉,约0.08时夏、秋线与春季线重合;前湖在划分阈值为0.06左右时夏季线与冬季线交叉,约0.08时夏季线与春、秋线交叉。也就是说,莫愁湖微生物群落Invsimpson指数在不同季节的排序表现为:当划分阈值<0.06时,秋>夏>冬>春;当0.06<划分阈值<0.08时,冬>夏≈秋>春;当划分阈值>0.08时,冬>夏≈秋≈春。同样,前湖数据中也存在这样的问题。可以看出,不同的划分阈值得到的样品微生物群落Invsimpson指数高低是不同的。而且图2d与图2c存在明显的一致性。
2.3讨论
由于很多序列目前还无法准确分类到属的水平,因此,按照属水平上的α多样性指数分析也存在一些缺陷。目前,对序列分析时常常会划分OTUs,0.03和0.1差异是常用的划分标准,大致对应科(family)和种(species)的分类学水平。但是由于不同的微生物类群之间rDNA/rRNA的变异速率是不同的,因此这样的分类也存在一定的缺陷。本研究从不同的角度来探讨南京莫愁湖、前湖、玄武湖、紫霞湖4个湖泊在不同季节微生物群落α多样性的变化,发现不同角度得出的结论是不同的。
1)OTUs和Chao1指数得出的结果基本一致,而Invsimpson与Shannon指数得出的结果也基本一致,这与它们属于不同的α多样性指数类别相关。OTUs和Chao1指数属于计算菌群丰度的指数,而Invsimpson与Shannon指数属于计算菌群多样性的指数。本研究中4个湖泊不同季节OTUs和Chao1指数随着OTUs划分阈值的递进,曲线不会出现交叉,表明划分阈值不会影响4个湖泊菌群丰度在不同季节的排序;而Invsimpson与Shannon指数随着划分阈值的递进,曲线出现交叉,表明划分阈值会影响菌群α多样性指数在不同季节的排序。
2)微生物群落组成差异较大时,可以得出基本一致的结论,如玄武湖:冬≈春>夏≈秋;但是当微生物群落组成差异较小时,得出的结论是不同的。比如,在属水平上前湖浮游细菌微生物群落α多样性:冬>春>秋>夏;从Invsimpson指数来看,当划分阈值<0.06时:夏>冬>春>秋,当0.06<划分阈值<0.08时:冬>夏>春≈秋,当划分阈值>0.08时:冬>春>秋>夏。这些结果似乎是互相矛盾的,因此,如果仅仅从一个方面来分析微生物群落的α多样性都是片面的,不能很好地解释样品的微生物群落组成多样性。
3)由表1可看出,在目前的技术下,依然无法准确判定大量的序列所对应的属水平,因此相较于样品之间已知属水平分类的序列占总序列的百分比相差很大时得出的结论的可靠性较低。本研究中发现莫愁湖已知属水平分类的序列占总序列的百分比在不同季节相差较小,在50%~60%之间,因此可以认为得到的不同季节在属水平微生物多样性是相对可靠的。属水平上莫愁湖在不同季节微生物多样性表现为:冬>夏≈秋>春,对比图2c、2d发现,在划分阈值大约在0.06~0.08时也有冬>夏≈秋>春,由此可以初步推断属水平所对应的OTUs划分阈值可能在0.06~0.08范围内。
4)一张图中α多样性指数曲线交叉点一般都出现在0.04~0.09,这表明OTUs划分阈值在一定区域内时对微生物α多样性影响很大,然而在这个区域外(<0.04或>0.09)对微生物α多样性的影响较小。因此,取划分阈值为0.03(< 0.04)和0.1(> 0.09)来划分是比较合理的。当α多样性指数出现交叉时,可以认为在不同的微生物分类学上的α多样性是不同的。比如,前湖Invsimpson指数在种的水平上:夏>冬>春>秋;然而在科的水平上:冬>春>秋>夏。
3结论
从多个方面探讨了南京莫愁湖、前湖、玄武湖、紫霞湖等4个湖泊浮游细菌群落的α多样性。通过比较OTUs划分阈值与α多样性指数曲线,从不同的分类学等级上研究了细菌群落的α多样性,为更好地理解微生物群落组成和α多样性奠定了基础。
参考文献:
[1]HE J Z,GE Y,XU Z H,et al.Linking soil bacterial diversity to ecosystem multifunctionality using backward elimination boosted trees analysis[J].Journal of Soils and Sediments,2009,9(6):547-554.
[2]SCHLOSS P D.A high-throughput DNA sequence aligner for microbial ecology studies[J].PLoS ONE,2009,4(12):e8230.
[3]SCHLOSS P D,WESTCOTT S L.Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis[J].Applied and Environmental Microbiology,2011,77(10):3219-3226.
[4]LEGENDRE P,LEGENDRE L F.Numerical Ecology[M].New York:Elsevier,1998.
[5]EDGAR R C.Search and clustering orders of magnitude faster than BLAST[J].Bioinformatics,2010,26(19):2460-2461.
[6]GORIS J,KONSTANTINIDIS K T,KLAPPENBACH J A,et al.DNA-DNA Hybridization values and their relationship to whole-genome sequence similarities[J].International Journal of Systematic and Evolutionary Microbiology,2007,57(1):81-91.
[7]HUGENHOLTZ P,GOEBEL B M,PACE N R.Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity[J].Journal of Bacteriology,1998,180(18):4765-4774.
[8]RAMETTE A,TIEDJE J M,KONSTANTINIDIS K T.The bacterial species definition in the genomic era[J].Philosophical Transactions of the Royal Society of London.Series B,Biological Sciences,2006,361(1475):1929-1940.
[9]CLEARY D F,SMALLA K,MENDONÇA-HAGLER L C,et al.Assessment of variation in bacterial composition among microhabitats in a mangrove environment using DGGE fingerprints and barcoded pyrosequencing[J].PLoS One,2012,7(1):e29380.
[10]GILLAN D C.The effect of an acute copper exposure on the diversity of a microbial community in North Sea sediments as revealed by DGGE analysis——the importance of the protocol[J].Marine Pollution Bulletin,2004,49(5):504-513.
[11]SCHLOSS P D,GEVERS D,WESTCOTT S L.Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies[J].PLoS ONE,2011,6(12):e27310.
[12]KUNIN V,ENGELBREKTSON A,OCHMAN H,et al.Wrinkles in the rare biosphere:Pyrosequencing errors can lead to artificial inflation of diversity estimates[J].Environmental Microbiology,2010,12(1):118-123.
[13]SCHLOSS P D.The effects of alignment quality,distance calculation method,sequence filtering,and region on the analysis of 16S rRNA gene-based studies[J].PLoS Computational Biology,2010,6(7):e1000844.
[14]HUSE S M,WELCH D M,MORRISON H G,et al.Ironing out the wrinkles in the rare biosphere through improved OTU clustering[J].Environmental Microbiology,2010,12(7):1889-1898.
[15]WANG Q,GARRITY G M,TIEDJE J M,et al.Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J].Applied and Environmental Microbiology,2007,73(16):5261-5267.
[16]SCHLOSS P D,HANDELSMAN J.Introducing DOTUR,a computer program for defining operational taxonomic units and estimating species richness[J].Applied and Environmental Microbiology,2005,71(3):1501-1506.
Effects of Different OTUs Cutoff Values onα-Diversity of Bacterioplankton in Lake
SHEN Feng1,2,HUANG Rui1,2,WANG Si-chen1,2,CAO Xin-yi1,2,ZHAO Da-yong1,2
(1.StateKeyLaboratoryofHydrology-WaterResourcesandHydraulicEngineering,HohaiUniversity,Nanjing210098,China;2.CollegeofHydrologyandWaterResources,HohaiUniversity,Nanjing210098,China)
Abstract:The α-diversity of bacterioplankton community in lake is generally evaluated based on OTUs (operation taxonomy units).This study investigated the effects of different OTUs cutoff values or genus level on α-diversity and detected the approximation of OTUs cutoff values which was corresponding to taxonomic level of genera for bacteria.By 454 high-throughput sequencing technology,the bacterioplankton communities of Mochou Lake,Qian Lake,Xuanwu Lake,and Zixia Lake in Nanjing city were studied.Different OTUs cutoff values were assumed to explore the variation of OTUs,Chao1,Invsimpson,Shannon indexes.Results showed that,OTUs and Chao1 indexes showed a consistent change with the variation of OTUs cutoff values,while Invsimpson and Shannon indexes showed a consistent change.Along with the distribution of OTUs cutoff values,OTUs and Chao1 indexes of four lakes remained monotonous compared to each other in different seasons,whereas the results of Invsimpson and Shannon indexes differed with fluctuated values.The taxonomic level of genera was roughly corresponding to differentiation threshold of 0.06~0.08.The well-known thresholds of 0.03 or 0.1 as cutoffs to analyze microbial sequence α-diversity was reasonable.Method for analyzing α-diversity of the microbial community was comprehensive and reliable.
Keywords:α-diversity;high-throughput sequence;OTUs;bacterioplankton;16S rRNA随着高通量测序技术的日渐流行和广泛使用,越来越多的稀有菌种被检测出来,这为微生物群落的深入研究提供了可能。而实际调查研究中,稀有细菌微生物的鉴定往往依赖于主观分类水平的选取,不同的种属划分等级亦导致微生物多样性计算结果的差异,而微生物多样性的变化对生态系统稳定性的影响极大[1]。
中图分类号:Q 93R 318
文献标识码:A
文章编号:1672-5425(2016)02-0025-06
doi:10.3969/j.issn.1672-5425.2016.02.005
作者简介:沈烽(1992-),男,江苏无锡人,硕士研究生,研究方向:湖泊环境微生物,E-mail:472779237@qq.com;通讯作者:赵大勇,教授,E-mail:dyzhao@hhu.edu.cn。
基金项目:国家自然科学基金资助项目(41371098,41571108),江苏省自然科学基金资助项目(BK20151614),中央高校基本科研业务费专项资金资助项目(2015B14214,2015B31714)
收稿日期:2015-11-13
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29 00:51:58
食品安全导刊(2021年20期)2021-08-30 06:40:50
制造技术与机床(2019年9期)2019-09-10 07:36:54
少儿美术(快乐历史地理)(2019年4期)2019-08-27 00:51:40
西南交通大学学报(2018年6期)2018-12-18 02:22:28
阅读(低年级)(2018年4期)2018-05-14 17:39:57
河北遥感(2017年2期)2017-08-07 14:49:00
小学阅读指南·低年级版(2017年2期)2017-03-23 13:07:24
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
水生生物学报(2015年1期)2015-02-28 16:01:05
