
ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
2016-04-08 06:35JieZhaoBingXuWenjieYuXuefengWangShanghaiKeyLabofChemicalAssessmentandSustainabilityDepartmentofChemistryTongjiUniversityShanghai200092China
Jie Zhao,Bing Xu,Wen-jie Yu,Xue-feng Wang∗Shanghai Key Lab of Chemical Assessment and Sustainability,Department of Chemistry,Tongji University,Shanghai 200092,China
ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
Jie Zhao,Bing Xu,Wen-jie Yu,Xue-feng Wang∗
Shanghai Key Lab of Chemical Assessment and Sustainability,Department of Chemistry,Tongji University,Shanghai 200092,China
(Dated:Received on November 16,2015;Accepted on December 30,2015)
The reaction of laser-ablated vanadium,niobium and tantalum atoms with hydrogen sul fi de has been investigated using matrix isolation FTIR and theoretical calculations.The metal atoms inserted into the H−S bond of H2S to form the HMSH molecules(M=V,Nb,Ta), which rearranged to H2MS molecules on annealing for Nb and Ta.The HMSH molecule can also further react with another H2S to form the H2M(SH)2molecules.These new molecules were identi fi ed on the basis of the D2S and H234S isotopic substitutions.DFT(B3LYP and BPW91)theoretical calculations are used to predict energies,geometries,and vibrational frequencies for these novel metal dihydrido complexes and molecules.Reaction mechanism for formation of group V dihydrido complex was investigated by DFT internal reaction coordinate calculations.The dissociation of HVSH gave VS+H2on broad band irradiation and reverse reaction happened on annealing.Based on B3LYP calculation releasing hydrogen from HVSH is endothermic only by 13.5 kcal/mol with lower energy barrier of 16.9 kcal/mol.
Key words:Hydrogen sul fi de,Matrix isolation,Transition metal,Density functional calculation
†Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
∗Author to whom correspondence should be addressed.E-mail: xfwang@tongji.edu.cn
I.INTRODUCTION
Abundant but toxic H2S from industry and nature is a potentially valuable chemical to produce clean hydrogen energy[1].However,it is a challenge to develop a sustainable and economical approach that can split H2S to H2and S simultaneously.The splitting reaction is thermodynamically unfavorable,which needs the standard Gibbs free energy change∆G−◦of 33 kJ/mol[2]. Up to now experiments have been done extensively for H2S activation[3–19],and the decomposition of H2S with photo catalysis has been explored[12,20,21].Hydrogen sul fi de also plays important roles in physiological processes such as vaso regulation and neutro transmission;for example,H2S molecule is an important signaling molecule that regulates the intracellular redox status and fundamental signaling processes at physiological levels[22].
The oxidation of H2S by transition metals has been studied both experimentally and theoretically[23–28]. Recent matrix isolation FTIR and theoretical study of reactions of laser-ablated thorium and uranium atom with H2S in excess noble gas matrixes has been reported [29].In solid argon group IV metal atoms insert into the S−H bond of hydrogen sul fi de to form the HMSH, H2MS,and H2M(SH)2(M=Ti,Zr,Hf)molecules[30]. In the case of group XII metal reactions only insertion products HMSH(M=Zn,Cd,Hg)were identifi ed.The HZnSH and HCdSH were obtained on sample annealing;however,the HHgSH was observed on UV irradiation[31].
In this work,we report here the identi fi cation of group V metal dihydrido complexes that are produced through laser-ablated V,Nb,Ta atom reactions with H2S in solid argon.The vibration frequencies are confi rmed by isotopic substitution and DFT frequency calculations.The metal dihydrido complexes observed in our experiment are instructive for designing the reversible hydrogen storage materials.
II.EXPERIMENTAL AND COMPUTATIONAL METHODS
Our experimental method has been described in detail previously[32–34].Brie fl y,a Nd:YAG laser fundamental(1064 nm,10 Hz repetition rate with 10 ns pulse width)was focused onto a rotating transition metal target through a hole in a CsI window cooled normally to 4 K by means of a closed-cycle helium refrigerator.The laser-evaporated metal atom was co-deposited with hydrogen sul fi de in excess argon onto the CsI window for 1 h at a rate of 2−4 mmol/h.The H2S,S,and D2S samples were prepared by previously described method [30].After deposition infrared spectra were recordedon a Bruker 80 V spectrometer at a 0.5 cm−1resolution between 4000 and 400 cm−1using a liquid nitrogen cooled broad band MCT detector.Matrix samples were annealed at di ff erent temperatures and were irradiated for 10 min by a mercury arc lamp(175 W)with globe removed to allow reagent di ff usion and further reaction.

FIG.1 Infrared spectra from the laser-ablated V atom reactions with H2S in excess argon.(a)V+0.5%H2S in argon codeposited for 1 h,(b)after annealing to 25 K,(c)after 300−740 nm irradiation,(d)after broad band irradiation,(e)after annealing to 30 K,(f)the laser ablated V atom reactions with 0.2%H2S+0.2%D2S in excess argon,after annealing to 25 K, (g)after 340−740 nm irradiation,(h)the laser ablated V atom reactions with 0.5%H234S in excess argon,after annealing to 25 K,(i)after 300−740 nm irradiation.
Quantum chemical calculations were performed using Gaussian 09 program soft package to help the assignment of vibrational frequencies of the observed reaction products[35].The B3LYP[36]and BPW91[37]density functional were applied and aug-cc-pVTZ-PP basis sets of Peterson et al.,which incorporate a relativistic pseudo potential that account for scalar relativistic e ff ects,were employed for Nb and Ta atoms[38,39]. The aug-cc-pVTZ basis sets were employed for V,S, and H atoms[40,41].The geometries of various reactants,intermediates,and products were fully optimized. The vibrational frequencies were calculated with analytic second derivatives,and zero-point energies were derived.The intrinsic reaction coordinate(IRC)calculations were carried out to ensure the obtained TSs connecting the desired reactants and products.
III.RESULTS AND DISCUSSION
A.Infrared spectra
Infrared spectra for the reactions of laser-ablated V,Nb,and Ta atoms with H2S in excess argon in the selected regions are illustrated in Fig.1−Fig.3, and the product absorptions are listed in Table I.In V−H stretching region absorptions were observed at 1682.8,1666.7,1630.3,and 1575.6 cm−1in experiment of vanadium atom reaction with H2S.The deuterium counterparts appeared at 1215.4,1206.0,1176.9,and 1136.1 cm−1in V+D2S experiment.In addition in V−S stretching region 583.5,562.9,and 529.4 cm−1bands appeared on 300−740 nm irradiation and enhanced greatly on broad band photolysis.The 583.5 cm−1band has been assigned to VS2molecule[43].Reactions of Nb atoms with H2S in excess argon produced new bands at 1726.9,1709.8,1695.6,and 1682.6 cm−1in Nb−H stretching region and 603.1 cm−1in Nb−S stretching region.Similarly Ta atom reactions in solid argon with H2S gave new absorptions at 1796.4,1780.7,1785.3, 1769.4,and 649.7 cm−1.For all three laser-ablated metal reactions experiments were also done with sulfur-34 isotopic-labeled samples(H234S)and deuterium labeled samples(H2S+HDS+D2S),and the representative spectra in selected region are shown in Fig.1−Fig.3, respectively.
B.Calculations
Calculations at the B3LYP level of theory were done for three isomers of MH2S,namely,the inserted HMSH and H2MS molecules,and the M(SH2)complexes.The calculated geometric parameters and relative stabilities of HMSH and H2MS isomers are shown in Fig.4,and the calculated vibrational frequencies and intensities are listed in Tables II−IV for observed molecules.We fi nd the HVSH molecule is the lowest in energy for all threeisomers;however,for Nb and Ta the dihydrido complex H2MS are the most stable on potential energy surface as shown in Fig.5.The HMSH and H2MS could react with H2S to give H2M(SH)2,which were calculated also to be dihydrido complex as shown in Fig.4.

FIG.2 Infrared spectra from the laser-ablated Nb atom reactions with H2S in excess argon;(a)Nb+0.5%H2S in argon code posited for 1 h,(b)after annealing to 30 K,(c)after 300−740 nm irradiation,(d)after broad band irradiation,(e) after annealing to 30 K,(f)the laser ablated Nb atom reactions with 0.2%H2S+0.2%D2S in excess argon,after annealing to 25 K,(g)after full-arc photolysis,(h)the laser ablated Nb atom reactions with 0.5%H234S in excess argon,after annealing to 25 K,(i)after broad band irradiation.
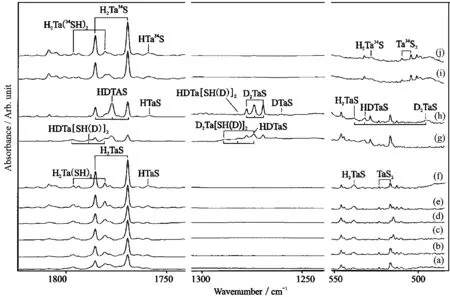
FIG.3 Infrared spectra from the laser-ablated Ta atom reactions with H2S in excess argon.(a)V+0.5%H2S in argon codeposited for 1 h,(b)after annealing to 25 K(c)after 300−740 nm irradiation,(d)after broad band irradiation,(e)after annealing to 30 K,(f)the laser ablated Ta atom reactions with 0.2%H2S+0.2%D2S in excess argon codeposited for 1 h, (g)after annealing to 25 K,(h)the laser ablated Ta atom reactions with 0.5%H234S in excess argon,codeposited for 1 h, (i)after annealing to 25 K.

TABLE I Infrared absorptions(cm−1)observed for products of the reaction of V,Nb,and Ta atoms with H2S molecules in argon.
C.Vanadium
1.HVSH
Laser-ablated vanadium atom reactions with H2S in excess argon gave the strongest new band at 1575.6 cm−1,which appeared on deposition and increased on 25 K annealing,but greatly decreased on broad-band photolysis.This band showed no sulfur-34 shift,but shifted to 1136.1 cm−1with D2S,giving an isotopic H/D ratio of 1.3868,suggesting that the 1575.6 cm−1band is due to V−H stretching mode.In the experiment with H2S+HDS+D2S mixture the doublet hydrogen isotopic distribution pattern(1575.6 and 1136.1 cm−1)was observed,con fi rming one hydrogen atom involved in this mode.So 1575.6 cm−1band is appropriate for the V−H stretching mode of HVSH molecule.Notice the V−H stretching frequency was observed for HV(OH)at 1567.0 cm−1in the vanadium atom reaction with H2O[42].
Theoretical calculations at B3LYP and BPW91 level predicted that the HVSH molecule with high-spin quartet ground state is in the global minimum energy (Fig.4).Our frequency calculation gave strong V−H stretching frequency at 1643.4 cm−1(B3LYP)and 1629.6 cm−1(BPW91),which is overestimated by 5.6% (B3LYP)and 3.4%(BPW91),respectively,in very good agreement with harmonic DFT frequency calculation for group V metal hydrides.In addition the calculated intensity of S−H stretching mode for HVSH is extremely weak(Table II),which is in agreement with our experiment in which this mode was not observed.
2.H2V(SH)2
Two weak bands at 1682.8 and 1666.7 cm−1appeared on initial deposition of vanadium atoms with H2S in excess argon,which increased upon annealing,but decreased on irradiation.These bands exhibited no sulfur-34 shift but shifted to 1215.4 and 1206.0 cm−1with deuterium substitution,giving H/D ratio of 1.3842 and 1.3833,respectively.The isotopic H/D ratios indicate that these two bands are due to V−H stretching vibrations.Experiment with H2S+HDS+D2S gave triplet distribution at 1682.8,1675.3,and 1666.6 cm−1in upper V−H stretching region and 1215.4,1211.1,and 1206.0 cm−1in lower V−D stretching region,suggesting two hydrogen atoms involved in the vibrations.These bands are assigned to V−H antisymmetric and symmetric modes of isolated H2V(SH)2molecule.
DFT frequency calculations substantiate the assignment of H2V(SH)2molecule.Firstly,with B3LYP functional the V−H stretching modes are computed for H2V(SH)2at 1809.1 and 1801.6 cm−1,respectively, which are overestimated by about 7%.These calculated results are in line with calculated and observed frequencies for other vanadium dihydrides[45].Secondly the calculated antisymmetric and symmetric V−H modes for H2V(SH)2are about 160 cm−1higher than the same modes for HVSH,which are in agreement with our observation.Thirdly,since the total energy of HVSH is 11 kcal/mol lower than that of H2VS that was not observed;however,in our experiment the major product HVSH further reacts with H2S to form H2V(SH)2and this reaction is exothermic by 22 kcal/mol based on our B3LYP calculation.In addition our BPW91 frequency calculations as listed in Table II also support the assignment.
3.HVS

FIG.4 B3LYP calculated geometric parameters(bond length in˚A)and relative stability(kcal/mol,in parentheses)of the MH2S(M=V,Nb,Ta)isomers and H2M(SH)2(M=V,Nb,Ta)molecules.
In V+H2S experiment a V−H stretching band at 1630.3 cm−1was observed on deposition and increased slightly on 300−740 nm irradiation.This band shows no shift with H234S sample but moved to 1176.9 cm−1 with D2S sample,giving H/D isotopic ratio of 1.3852. A weak band at 562.9 cm−1tracked with 1630.3 cm−1 in the whole reaction process.S and D2S,the counterpart bands,appeared at 556.1 and 554.7 cm−1, de fi ning32S/34S and H/D ratio of 1.0122 and 1.0148, respectively.The large34S shift indicates this absorption arises mostly from the V−S stretching vibration perturbed by VH vibrations.A new triatomic HVS molecule is proposed and con fi rmed by DFT calculations.With B3LYP functional frequency calculation the M−H stretching mode of the HVS molecules was calculated at 1719.8 cm−1,which is overestimated by 5.5%.In addition M−S stretching modes was predicted at 577.8 cm−1that is in very good agreement with observation.
4.VS and VS2
In the V+H2S experiment a weak band in V−S stretching region at 529.4 cm−1is due to diatomic VS molecule,which was observed on deposition and greatly enhanced on broad band photolysis at the expense of HVSH and H2V(SH)2molecules.This band exhibited no deuterium isotopic shift but shifted to 519.7 cm−1 withS,and gave an isotopic32S/34S ratio of 1.0187.S mixture(not shown)doublet distribution at 529.4 and519.7 cm−1was observed and assignment of VS is con fi rmed.Notice this band was observed weakly in early vanadium atom reaction with discharged sulfur vapor[43].Another band at 583.5 cm−1appeared on 300−740 nm irradiation and enhanced greatly on broad-band photolysis,which has been assigned to VS2molecule[43].The V−S stretching modes for VS and VS2molecules were calculated at 539.8 and 593.0 cm−1 (B3LYP),respectively,which are overestimated by only 10 cm−1.The calculated BPW91 frequencies are even better matching observed values(Table II).
D.Niobium
1.H2NbS and H2Nb(SH)2
The absorptions at 1709.8,1682.8 and 603.4 cm−1in the Nb+H2S experiments are assigned to the H2NbS molecule.These three bands increased on annealing and decreased on broad-band irradiation.The 1709.8 and 1682.8 cm−1bands show no sulfur-34 shift withS sample,but with deuterium enriched sample these bands shift to 1226.7 and 1210.1 cm−1,defying H/D ratios of 1.3938 and 1.3906,respectively,which are typical Nb−H stretching vibrations.In the mixed H2S+HDS+D2S experiments,two intermediate bandswere observed at 1696.1 and 1218.6 cm−1;which clearly show that two equivalent H atoms are involved in this molecule.The 603.4 cm−1band shifted to 598.3 cm−1 withS and to 563.3 cm−1with D2S,giving32S/34S ratio of 1.0085 and H/D ratio of 1.0711,respectively. An intermediate band at 582.2 cm−1appeared in the mixed H2S+HDS+D2S experiments.Accordingly,the 1709.8 and 1682.8 cm−1bands are assigned to the Nb−H symmetry and anti-symmetry stretching vibrations and the 603.4 cm−1band is assigned to NbH2bending vibration coupled with the NbS vibration in H2NbS molecule.
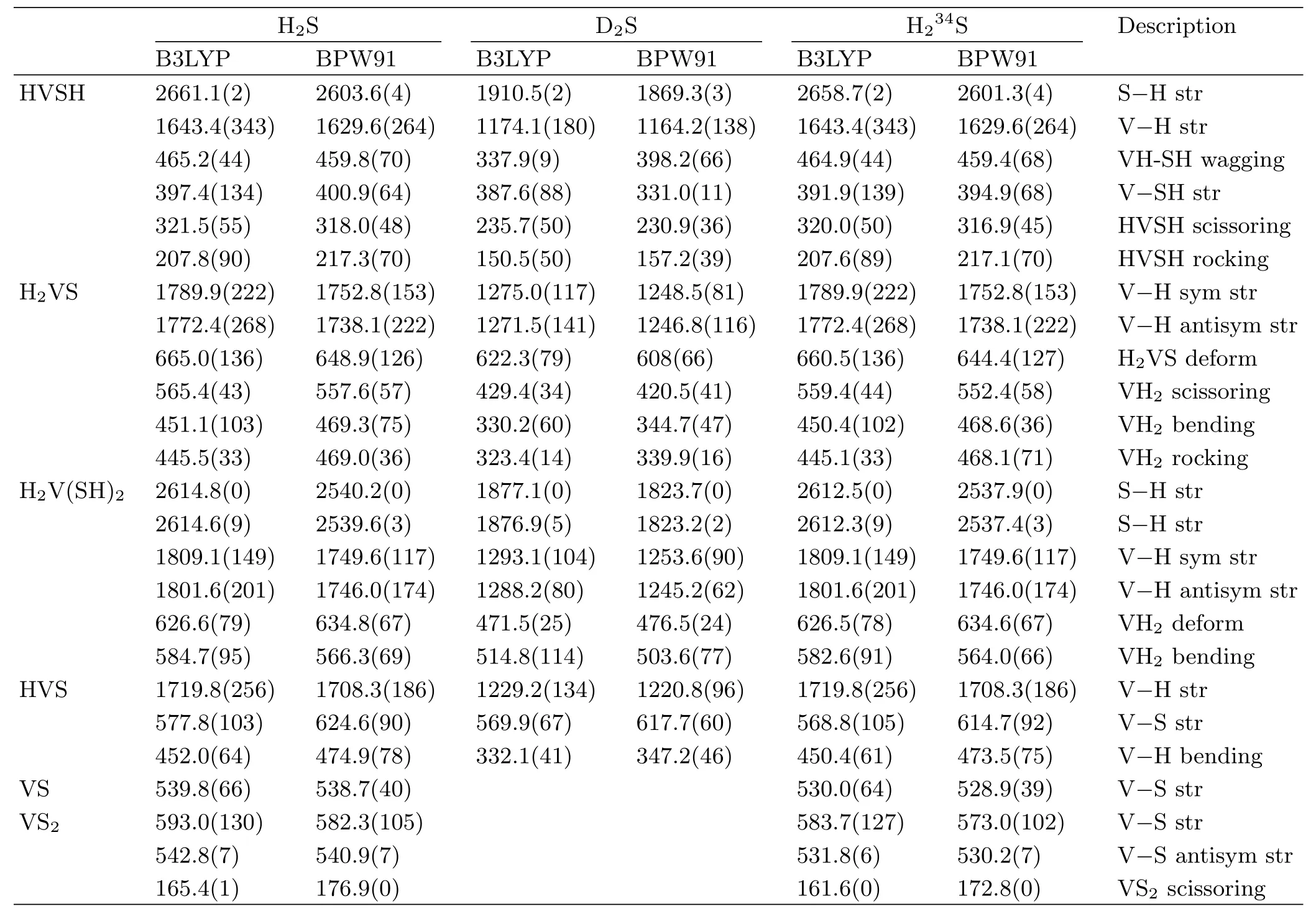
TABLE II Calculated frequencies for HVSH,H2VS,H2V(SH)2,HVS,VS,and VS2.
In the reactions of atomic Nb with H2S in solid argon,absorptions at 1726.9 and 1695.6 cm−1track together,which are located in Nb−H stretching region. These bands are weak on initial deposition and increasing greatly on annealing to 25 K and further increasing by 50%on annealing to 30 K.WithS there were no shifts observed but with deuterium substitution two bands shifted to 1249.2 and 1218.8 cm−1,de fi ning 1.3824 and 1.3912 H/D ratios,respectively.In the mixture H/D isotopic experiment(H2S+HDS+D2S)two triplets were observed in upper Nb−H and lower Nb−D stretching regions.These bands favored on later annealing,suggesting the product comes from niobium atom reaction with more H2S molecules.Accordingly the H2Nb(SH)2molecule is proposed.
The B3LYP calculation predicted that the H2NbS has a2A′ground state and NbH symmetric and antisymmetric stretching modes are calculated at 1787.3 and 1772.6 cm−1,which are overestimated by 4.5% and 5.3%,respectively.The predicted H-Nb−S bending mode at 679.1 cm−1shows 2 cm−1shift with S-34 substitution and 97 cm−1shift with deuterium substitution,which match the observed values very well.Our BPW91 frequency computation predicted the Nb−H symmetric and antisymmetric stretching modes of H2Nb(SH)2at 1761.6 and 1750.3 cm−1,which are overestimated by 3.0%and 4.0%,respectively.
2.NbS and NbS2
In the Nb+H2S experiments,a weak band at 541.2 cm−1observed on deposition increased 50%on 300−700 nm irradiation.This band showed no deuterium shift but shifted to 533.9 cm−1withS sample,giving 1.0137 isotopic ratio,which is appropriate for diatomic NbS molecule.A band at 531.1 cm−1appeared on co-deposition and increased on broadband photolysis,and shifted to 520.4 cm−1with H234S sample,de fi ning the32S/34S isotopic ratio of 1.0206. This band has been assigned to NbS2molecule[43]. The NbS stretching mode is predicted at 539.4 cm−1, which is overestimated by 1.5%.Notice the increase of 541.2 cm−1band is at expense of H2NbS on UV irradiation.

TABLE III Calculated frequencies for HNbSH,H2NbS,H2Nb(SH)2,HNbS,NbS,and NbS2.
E.Tantalum
1.H2TaS and H2Ta(SH)2
In solid argon,the reactions of Ta with H2S gave a set of bands at 1785.5,1769.4 and 538.3 cm−1, which tracked together in the whole reaction process. These bands appeared on the co-deposition,increased two folds on 25 K annealing,slightly increased on 300−740 nm irradiation,but decreased on broad band irradiation.The 1785.5 and 1769.4 cm−1bands showed no34S shift but moved to 1277.6 and 1269.4 cm−1with D2S sample,de fi ning the 1.3975 and 1.3935 H/D ratios. The 538.3 cm−1band shifted to 528.5 cm−1with H234S de fi ning the isotopic32S/34S ratio of 1.0187,and the deuterium counterpart was observed at 495.6 cm−1,giving H/D ratio of 1.0862.Using a mixed H2S+HDS+D2S sample,three triplet band distributions were observed in upper Ta−H stretching region at 1785.5,1777.1,and 1769.4 cm−1,lower Ta−D stretching region at 1277.6, 1274.0,and 1269.4 cm−1,and Ta-S stretching region at 538.3,531.7 and 495.6 cm−1,indicating two H atoms are involved in the molecule.On the basis of the above spectroscopic information,this set of bands is assigned to the H2TaS molecule.Notice the 1777.1,1274.0 and 531.7 cm−1bands are due to Ta−H and Ta−D stretching,and TaHD bending vibrations of HDTaS,respectively.
The 1796.4 and 1780.7 cm−1absorptions in the Ta+H2SexperimentsareassignedtoH2Ta(SH)2molecule.Two bands increased on annealing to 25 K, slightly decreased on full-arc photolysis and increased again on annealing to 30 K.These two bands showed no sulfur-34 shift with H234S sample and the deuterium counterparts were found at 1288.7 and 1277.6 cm−1, giving 1.3940 and 1.3938 H/D ratios.Two triplet distributions appeared in upper Ta−H and lower Ta−D stretching regions in the mixed H2S+HDS+D2S experiments,indicating two equivalent H atoms included inthe molecule.Accordingly these bands were assigned to H2Ta(SH)2molecule.

TABLE IV Calculated frequencies for HNbSH,H2NbS,H2Nb(SH)2,HNbS,NbS,and NbS2.
B3LYPcalculationpredictedthattheH2TaS molecule has a2A′ground state with nonplanar geometry.The Ta−H stretching and HTaH bending vibrations were calculated at 1845.7,1829.1,and 547.0 cm−1, respectively,which require scaling factor of 0.967,0.967, and 0.984.The calculated Ta−H symmetric and antisymmetric stretching modes of H2Ta(SH)2are at 1862.2 and 1844.9 cm−1which match the observed values very well.Notice our BPW91 frequency calculations also support band assignments for H2TaS and H2Ta(SH)2molecules.
F.Other absorptions
In Nb+H2S experiment a new Nb−H mode at 1672.7 cm−1appeared on visible light irradiation and shifted to 1198.8 with D2S sample.Similarly in the Ta+H2S experiment a new Ta−H stretching frequency at 1759.0 cm−1was observed,which shifted to 1260.5 cm−1,defying 1.3955 H/D ratio.The triatomic molecule HMS(M=Zr,Hf)comes into mind for these band assignment though the M−S stretching frequencies are missing.The M−H stretching modes of the HMS molecules were calculated at 1759.3 cm−1 (Nb)and 1825.3 cm−1(Ta)with B3LYP calculation, which are overestimated by 5.4%and 3.8%,respectively. In addition M−S stretching modes were predicted at 548.8 cm−1(Nb)and 553.1 cm−1(Ta).Unfortunately these modes were not observed in our experiments.
G.Reaction mechanism


FIG.5 Potential energy surface for reaction of V,Nb,Ta with H2S at B3LYP level of theory.
As shown in Fig.5,the formations of VSH2(4A′), NbSH2(4A′),and TaSH2(4A′)complexes from metal atom and hydrogen sul fi de are calculated exothermically by 5.8,13.0,and 5.2 kcal/mol,respectively.These complexes are extremely reactive,which rearrange to insertion products HMSH by hydrogen shift from sulfur atoms to metal atoms exothermically by 44.0,53.5,and 52.5 kcal/mol for HVSH,HNbSH,and HTaSH.Next, the second hydrogen atom on sulfur atom of the insertion products can shift to metal atom to produce the dihydrido complex H2MS. It should be noted that the H2VS(2A′)is 11.1 kcal/mol higher in energy than HVSH,indicating the formation of HVSH is thermodynamically favored,which is in accord with our observation that main reaction product is HVSH in the experiment of V+H2S in argon matrix. However for Nb and Ta,dihydrido complex H2MS is more stable.H2NbS(2A′)and H2TaS(2A′)are 24.9 and 39.2 kcal/mol lower in energy than HNbSH(4A′)and HTaSH(4A′),respectively.In Nb and Ta reactions with H2S,the H2NbS and H2TaS were observed as majorproducts.Notice there is spin crossing to form these molecules from HMSH but in low temperature matrix spin crossing has high e ffi ciency since the4A′and2A′states of HNbSH and HTaSH lies very close in energy.
The diatomic VS molecule increased on photolysis and decreased on annealing with HVSH behavior on the contrary way in V+H2S experiment,indicating the dissociation of HVSH gave VS+H2,and reverse reaction happened on annealing.Compared with the reaction of V with H2O in argon matrix reported by Zhou et al.,where the release of hydrogen from HVOH is endothermic by about 17.3 kcal/mol with a barrier as high as 35.8 kcal/mol based on theoretical calculation[42]. However,releasing hydrogen from HVSH is endothermic only by 13.5 kcal/mol with lower energy barrier of 16.9 kcal/mol,indicating vanadium sul fi des may be a better hydrogen energy storage material than vanadium oxides as the hydrogen storage and release process are easier to take place.Metal sul fi des such as MoSχand BiSχhave potential applications for hydrogen storage because of their unique characteristics of strong gas adsorptions[44].
Furthermore,the insertion products HMSH can also react with H2S to give H2M(SH)2on annealing.

The H2M(SH)2molecules were decomposed into hydrogen and MS2under broad-band photolysis,which are endothermic by 16.3,19.5,and 22.4 kcal/mol,respectively.

IV.CONCLUSION
The reactions of laser-ablated V,Nb,and Ta atoms with sulfur hydride have been investigated using matrix isolation FTIR and theoretical calculations.The V,Nb, and Ta atoms inserted into the H−S bond of sulfur hydride to form the HMSH molecules(M=V,Nb,Ta), which further rearrange to H2MS molecules on annealing for Nb and Ta.HMSH can also further react with other sulfur hydride to form the H2M(SH)2molecules. The molecules were identi fi ed on the basis of the D2S and H234S isotopic substitutions as well as density functional calculations.Qualitative analysis of the reaction paths leading to the observed products was proposed. The H2elimination process was observed for group V metal dihydrido complexes on broad band irradiation.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.21173158 and No.21373152)and the Ministry of Science and Technology of China(No.2012YQ220113-7).
[1]J.Zaman and A.Chakma,Fuel Process.Technol.41, 159(1995).
[2]H.Shiina,M.Oya,K.Yamashita,A.Miyoshi,and H. Matsui,J.Phys.Chem.100,2136(1996).
[3]T.Chivers,J.B.Hyne,and C.Lau,Int.J.Hydrogen Energy 5,499(1980).
[4]E.A.Fletcher,J.E.Noring,and J.P.Murray,Int.J. Hydrogen Energy 9,587(1984).
[5]J.F.Reber and K.Meier,J.Phys.Chem.88,5903 (1984).
[6]D.W.Kalina and E.T.Maas Jr.,Int.J.Hydrogen Energy 10,163(1985).
[7]D.W.Kalina and E.T.Maas Jr.,Int.J.Hydrogen Energy 10,157(1985).
[8]J.F.Reber and M.Rusek,J.Phys.Chem.90,824 (1986).
[9]L.M.Al-Shamma and S.A.Naman,Int.J.Hydrogen Energy 15,1(1990).
[10]K.Petrov and S.Srinivasan,Int.J.Hydrogen Energy 21,163(1996).
[11]S.V.Tambwekar and M.Subrahmanyam,Int.J.Hydrogen Energy 22,959(1997).
[12]Z.Lei,W.You,M.Liu,G.Zhou,T.Takata,M.Hara, K.Domen,and C.Li,Chem.Commun.214(2003).
[13]H.Wang,Int.J.Hydrogen Energy 32,3907(2007).
[14]G.B.Zhao,S.John,J.J.Zhang,J.C.Hamann,S.S. Muknahallipatna,S.Legowski,J.F.Ackerman,and M. D.Argyle,Chem.Eng.Sci.62,2216(2007).
[15]H.Huang,Y.Yu,and K.H.Chung,23,4420(2009).
[16]T.Nunnally,K.Gutsol,A.Rabinovich,A.Fridman, A.Starikovsky,A.Gutsol,and R.W.Potter,Int.J. Hydrogen Energy 34,7618(2009).
[17]H.Yan,J.Yang,G.Ma,G.Wu,X.Zong,Z.Lei,J. Shi,and C.Li,J.Catal.266,165(2009).
[18]E.Linga Reddy,V.M.Biju,and C.Subrahmanyam, Appl.Energy 95,87(2012).
[19]X.Zong,J.Han,B.Seger,H.Chen,G.Lu,C.Li,and L.Wang,Angew.Chem.Int.Ed.53,4399(2014).
[20]G.Ma,H.Yan,X.Zong,B.Ma,H.Jiang,F.Wen,and C.Li,Chin.J.Catal.29,313(2008).
[21]G.Ma,H.Yan,J.Shi,X.Zong,Z.Lei,and C.Li,J. Catal.260,134(2008).
[22]F.Yu,X.Han,and L.Chen,Chem.Commun.50,12234 (2014).
[23]A.J.Tursi and E.R.Nixon,J.Chem.Phys.53,518 (1970).
[24]R.R.Smardzewski and M.C.Lin,J.Chem.Phys.66, 3197(1977).
[25]E.L.Woodbridge,T.L.Tso,M.P.McGrath,W.J. Hehre,and E.K.C.Lee,J.Chem.Phys.85,6991 (1986).
[26]J.D.Carpenter and B.S.Ault,J.Phys.Chem.96, 7913(1992).
[27]E.Isoniemi,M.Pettersson,L.Khriachtchev,J.Lundell, and M.R¨as¨anen,J.Phys.Chem.A 103,679(1999).
[28]S.J.Thompson,N.Goldberg,and B.S.Ault,PCCP 8,856(2006).
[29]X.Wang,L.Andrews,K.S.Thanthiriwatte,and D.A. Dixon,Inorg.Chem.52,10275(2013).
[30]Q.Wang,J.Zhao,and X.Wang,J.Phys.Chem.A (2014).
[31]Z.Pan,X.Liu,J.Zhao,and X.Wang,J.Mol.Spectrosc.310,16(2015).
[32]L.Andrews,X.Wang,Y.Gong,G.P.Kushto,B. Vlaisavljevich,and L.Gagliardi,J.Phys.Chem.A 118, 5289(2014).
[33]L.Andrews,Chem.Soc.Rev.33,123(2004).
[34]X.Liu,X.Wang,B.Xu,and L.Andrews,PCCP 16, 2607(2014).
[35]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E.Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani,V. Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji, M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov, J.Bloino,G.Zheng,J.L.Sonnenberg,M.Hada,M. Ehara,K.Toyota,R.Fukuda,J.Hasegawa,M.Ishida, T.Nakajima,Y.Honda,O.Kitao,H.Nakai,T.Vreven, J.A.Montgomery Jr.,J.E.Peralta,F.Ogliaro,M. Bearpark,J.J.Heyd,E.Brothers,K.N.Kudin,V. N.Staroverov,T.Keith,R.Kobayashi,J.Normand, K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo, J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski, R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,¨O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski,and D.J.Fox,Gaussian 09,Revision C.01, Wallingford CT:Gaussian,Inc.,(2010).
[36]C.Lee,W.Yang,and R.G.Parr,Phys.Rev.B 37,785 (1988).
[37]J.P.Perdew and Y.Wang,Phys.Rev.B 45,13244 (1992).
[38]K.A.Peterson,D.Figgen,M.Dolg,and H.Stoll,J. Chem.Phys.126,(2007).
[39]D.Figgen,K.A.Peterson,M.Dolg,and H.Stoll,J. Chem.Phys.130,(2009).
[40]D.E.Woon and T.H.Dunning,J.Chem.Phys.100, 2975(1994).
[41]D.E.Woon and T.H.Dunning,J.Chem.Phys.98, 1358(1993).
[42]M.Zhou,J.Dong,L.Zhang,and Q.Qin,J.Am.Chem. Soc.123,135(2000).
[43]B.Liang and L.Andrews,J.Phys.Chem.A 106,3738 (2002).
[44]C.H.Lai,M.Y.Lu,and L.J.Chen,J.Mater.Chem. 22,19(2012).
[45]X.F.Wang and L.Andrews,J.Phys.Chem.A 115, 14175(2002).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study†
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study†
- ARTICLE Infrared Photodisssociation Spectroscopy of Boron Carbonyl Cation Complexes†