
REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
2016-04-08 06:35WenkaiZhangCenterforAdvancedQuantumStudiesDepartmentofPhysicsBeijingNormalUniversityBeijing100875China
Wen-kai Zhang∗Center for Advanced Quantum Studies,Department of Physics,Beijing Normal University,Beijing 100875,China
REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
Wen-kai Zhang∗
Center for Advanced Quantum Studies,Department of Physics,Beijing Normal University,Beijing 100875,China
(Dated:Received on December 1,2015;Accepted on December 28,2015)
Polarization dependent time-resolved infrared(TRIR)spectroscopy has proven to be a useful technique to study the structural dynamics in a photochemical process.The angular information of transient species is obtainable in this measurement,which makes it a valuable technique for the investigation of electron distribution,molecular structure,and conformational dynamics.In this review,we brie fl y introduce the principles and applications of polarization dependent TRIR spectroscopy.We mainly focused on the following topics:(i)an overview of TRIR spectroscopy,(ii)principles of TRIR spectroscopy and its advantages compared to the other ultrafast techniques,(iii)examples that use polarization dependent TRIR spectroscopy to probe a variety of chemical and dynamical phenomena including protein conformational dynamics,excited state electron localization,and photoisomerization,(iv)the limitations and prospects of TRIR spectroscopy.
Key words:Ultrafast spectroscopy,Infrared spectroscopy,Polarization,Time-resolved infrared spectroscopy
†Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
∗Author to whom correspondence should be addressed.E-mail: wkzhang@bnu.edu.cn
I.INTRODUCTION
Femtosecond resolution studies of photochemical dynamics have the potential to detect the critical nuclear motions in real time from which a reaction mechanism can be constructed,understood,and ideally controlled[1−5].After the ultrashort UV/visible pulse excites a molecule,the subsequent evolution of the excited species can be followed by time-resolved fl uorescence[6−8],transient absorption in the UV/visible and infrared regions[9−11],X-ray di ff raction[12−14]and spectroscopy[15−18],and electron di ff raction[19,20]. Even though X-ray and electron probe can provide more insights into the structural dynamics,the technical diffi culties limit them to very few labs and research facilities and prevent their accessibility to the larger research community.On the other hand,transient electronic absorption spectroscopy has been intensively used to study the ultrafast dynamics in chemistry,physics and biology for decades.But electronic spectroscopy is typically very broad and relatively featureless,which makes structure determination extremely challenging.However,vibrational spectroscopy can identify the absorbing species more precisely than electronic spectroscopy because the absorption bands of vibrational transitions are narrower and less overlapped.Furthermore,because vibrational transitions are more spatially localized than electronic transitions,time-resolved vibrational spectroscopy can provide more insights into the structural dynamics[21−23].In the case where speci fi c vibrational modes correlate with speci fi c vibrational motions, one can directly obtain a structural information of the photo-induced reaction by inspecting the changes in vibrational absorption.Two primary vibrational spectroscopy methods,time-resolved Raman[24−29]and infrared spectroscopy[30−37],have been extensively used to study photochemical dynamics.
When the vibrational mode of interest is a local mode, one can directly link the transition dipole moment with the particular chemical bond that is modulated by the vibration.For instance,in the fi rst notable application of polarization dependent time-resolved infrared (TRIR)spectroscopy,Hochstrasser and coworkers examined the orientation of bound CO to myoglobin by detaching CO from carboxy myoglobin(MbCO)with polarized laser pulses and investigated its recombination to the active center by infrared absorption[38]. The authors extracted the angle of the CO in the protein frame of MbCO by monitoring the bleaching signal under di ff erent excitation conditions.They concluded that the Fe−C bond tilts to the heme normal and the Fe−C−O angle di ff ered signi fi cantly from 180◦.Recently,An fi nrud and coworkers showed that this deviation is less than 7◦by carefully controlling the experimental variables[39].Since then,polarization dependent TRIR spectroscopy has been extensively utilizedto reveal the orientational dynamics of the CO and NO ligands in myoglobin and hemoglobin,either bound to the heme iron or in the heme pockets[40−46].It is worth noting that the assumption that the transition dipole moment lies along the CO bond vector is incorrect in most cases.
Green fl uorescent protein(GFP)and its chromophore 4′-hydroxybenzylidene-2,3-dimethylimidazolinone (HBDI)provide another notable example.The HBDI chromophore in solution has an excited state lifetime of 1.2 ps and a fl uorescence quantum yield of only 10−3 [47−49]while the same chromophore has a fl uorescence lifetime of~3 ns and quantum yield approaching 0.8 in wild-type GFP[50−53].Theoretical calculations suggested that the fl exibility of room temperature solvent leads to bond isomerization and ultrafast excited state quenching for the HBDI chromophore [54].Usman et al.measured the TRIR anisotropy of a localized CO stretching mode in HBDI[55]. They observed an upshifted broadband(with fwhm of~50 cm−1)excited state absorption(ESA)feature at~1750 cm−1in natural HBDI.Their experiment indicated a di ff erent excited state behavior from the formation of a charge transfer state,provided an angle of 70◦between the electronic transition dipole moment and CO vibrational transition dipole moment,and concluded that structural change in HBDI is due to an isomerization by a single twist or a hula twist [55].Later,van Thor and coworkers showed that the HBDI isomerization is signi fi cantly reduced in the GFP excited state due to the constraint of the protein environment[52].There are more examples that employed TRIR anisotropy measurements including the isomerization reaction of photoactive yellow protein (PYP)[56,57],phytochrome holoprotein[58],and the structural response of an enzyme to a photo-excited inhibitor[59].It has also been used to determine the three-dimensional orientation of electronic transition dipole moment[60−62]and to follow the photo-induced transfer dynamics and the structural evolution of the charge separated states[63,64].
Even though the TRIR spectroscopy has been extensively used to study the structural dynamics of the photochemistry process,the di ffi culties in robustly interpreting femtosecond resolution measurements reduce its ability to determine photochemical reaction mechanisms.As a consequence,the TRIR spectroscopy and its application in the chemical reaction dynamics have been underutilized.Within this context,we believe there is a need to introduce the principle of polarization dependent TRIR spectroscopy and summarize its applications in structural dynamics to a larger research community.In this review,we fi rst give an overview of the TRIR spectroscopy and describe its advantages in resolving structural dynamics.We then present examples of using anisotropy measurements to study electron localization dynamics in charge transfer excited states and bond isomerization dynamics in a push-pull donorphenyl-accepter system.We show that the recent improvements in ultrafast laser technology,continuous advances in experimental methodology,and the advent of a common language for the interpretation of measurements,have assisted the study of chemical dynamics. We then brie fl y describe the challenges of ultrafast infrared spectroscopy in biological applications and conclude with a future outlook.
II.PRINCIPLES OF POLARIZATION DEPENDENT TRIR SPECTROSCOPY
The TRIR spectroscopy employs a pump-probe methodology,where the pump pulse is typically in the UV/visible region and the probe pulse is in the midinfrared(mid-IR)region for most photochemistry studies.Femtosecond mid-IR pulses are generated through a di ff erence frequency generation process after optical parametric ampli fi er[65].Experimentally,TRIR spectroscopy is performed in a spectrally-resolved con fi guration.The pump-induced absorbance changes are then measured with a mercury cadmium telluride(MCT)detector after spectral dispersion using a monochromator. A side e ff ect is that the ground state bleach(GSB)signals appear to grow at negative time delay,which is the so-called perturbed free induction decay.It is a common feature when the dephasing time of bleached transitions is much longer than the cross-correlation time between the pump and probe pulse,which is about 100−200 fs [66−68].
The signal contribution in the UV/visible pump mid-IR probe TRIR spectroscopy is much simpler than conventional transient absorption spectroscopy.As we know,there are three distinct sources of signals in the transient absorption measurements as illustrated by the energy diagram in Fig.1(a).The GSB and stimulated emission(SE)lead to an increase in signal transmission while the ESA reduces the signal transmission. The potential spectral overlap between GSB,ESA,and SE prevents the application of the transient absorption spectroscopy to many interesting problems,and the situation can be even worse in the transition metal related systems.However,there is no SE contribution in UV/visible pump mid-IR probe TRIR spectroscopy since the frequency of the mid-IR probe is signi fi cantly lower than the pump frequency and the possible electronic excited state emission as shown in Fig.1(b),which will remarkably reduce the di ffi culty in distinguishing the signal from di ff erent contributions.
Polarization dependent TRIR spectroscopy measures the frequency dependent isotropic,Iiso(ω,t),and the anisotropic,r(ω,t),signals from the parallel and perpendicular polarization measurements[69],


where I‖and I⊥represent the changes in probe transmission induced by a pump pulse when the pump and probe pulses have parallel and perpendicular polarizations.The experimental and theoretical framework developed for the conventional polarization dependent time-resolved spectroscopy method can be easily applied to the polarization dependent TRIR spectroscopy [70−79].For example,the TRIR anisotropy provides insights into the relative angles between the electronic and infrared transition dipole moments[80,81].Under the most common circumstances,a low concentration of chromophores with non-degenerate excited states will have an anisotropy that ranges from−0.2 to 0.4,where the decay of the anisotropy results from the rotation of excited state molecules.In next section,we will present a detailed example of using TRIR anisotropy measurement to study electron localization dynamics in charge transfer excited states.
III.ELECTRON LOCALIZATION IN CHARGE TRANSFER EXCITED STATES
E ffi cient energy migration and charge separation are essential steps in molecularly based light-harvesting materials[82,83].Charge transfer excited states of a high symmetry coordination complex have either an electron or a hole residing in one of the degenerate molecular orbitals.For the idealized degenerate case,the coupling between degenerate molecular orbitals leads to delocalization of the excited state,while static and dynamic disorder will reduce the symmetry and eliminate the energetic degeneracy that provide a mechanism for electron localization.For these reasons,the time-dependent charge transfer excited states in high symmetry coordination complexes provide a particular example of assessing how fundamental molecular properties control excited state electronic structure and charge separation.
A lot of experimental and theoretical studies have emphasized the importance of time-resolved anisotropy in the structural dynamics of the electronic excited state for high symmetry molecules[84−89].When the pump pulse excites degenerate states,the initial value of the anisotropy re fl ects the molecular symmetry and the decay of the anisotropy re fl ects multiple dynamical processes.Molecules with three-fold degeneracy will have initial anisotropy r(0)=1.0 while molecules with twofold degeneracy will have initial anisotropy r(0)=0.7 [85].A schematic of the relevant processes for twofold degenerate system appears in Fig.2.The dynamical phenomena that govern the loss of anisotropy for the two-fold degenerate states can be expressed as three rates re fl ecting three distinct processes for an overdamped superposition of excited states[89],

Dephasing due to inter-and intra-molecular fl uctuations occurs with a γ rate and leads to localization of the charge transfer excited state to a single molecular orbital and reduction in anisotropy to r=0.4.Incoherent electron transfer between degenerate localized charge transfer excited states occurs with a Γ rate and further reduces the anisotropy to r=0.1 for two-fold degenerate states.Excited state bond rotation can also lead to changes in the anisotropy[90],though not for the charge transfer systems discussed in this case.For dilute excitation,where excitation transfer between molecules does not occur,molecular rotation with the rate D causes the fi nal loss of anisotropy.
The contradictory interpretations of anisotropy measurementsforthemetal-to-ligandchargetransfer (MLCT)excited state of ruthenium-tris-bipyridine highlight the challenges in interpreting time-resolved anisotropy results[86−88].The photoexcitation leads to two-fold degenerate electronic excited states with orthogonal transition dipole moments.The biggest problem for anisotropy measurement occurs when the ESA spectrally overlaps with either the GSB or the SE. When signals of opposite sign spectrally overlap,the anisotropy extracted from Eq.(2)can range from−∞to+∞which makes the measurement meaningless[88]. Here we show that these di ffi culties in experimental interpretation can be partially addressed by a change in experimental design.We have used polarization dependent transient mid-IR absorption spectroscopy to study the electron localization dynamics of Fe(CN)63−[69]. Using the TRIR spectrum of CN-stretch vibration to track the dynamics of electronic excited states has the following advantages.The simplicity of CN-stretch vi-brational lineshapes allow us to distinguish clearly the ESA from the GSB signal,and the recorded transient vibrational spectrum does not have an SE contribution. The CN-stretch modes have transition dipole moments parallel to the CN bonding axes in this system,which greatly simpli fi es the interpretation of the anisotropy measurements.

FIG.2 A sketch of the electronic excited state relaxation processes for a two-fold degenerate system.γ is the rate of the decoherence,Γ is the rate of the incoherent electron transfer,and D is the rate of molecular rotation.This fi gure is adapted with permission from Zhang et al.[18],copyright(2015),American Chemical Society.
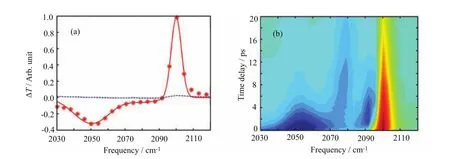
FIG.3 TRIR spectroscopy for[Fe(CN)6]3−dissolved in dimethyl sulfoxide.(a)The isotropic(red points)and S‖−S⊥di ff erence(blue traces)transient spectra are shown for a 0.2 ps time delay.The solid red line is the fi t of isotropic transient spectra.(b)Isotropic transient spectra as a function of mid-IR probe time delay and frequency.These fi gures are adapted with permission from Zhang et al.[18],copyright(2012)American Chemical Society.
We generate a ligand to metal charge transfer (LMCT)excited state and probe the electronic excited state dynamics with mid-IR pulses polarized parallel and perpendicular to the UV pump polarization.The octahedral Fe(CN)63−complex only has a three-fold degenerate T1uCN stretch mode in the mid-IR region.As shown in Fig.3(a),strong inter-ligand electronic coupling in the LMCT excited state preserves the octahedral symmetry and leads to a single T1uCN-stretch ESA band at 2050 cm−1with no anisotropy by a 0.2 ps time delay.With a solvent dependent rate,we observed this ESA converts to two ESA peaks appearing at 2079 and 2095 cm−1with a 5 ps time constant as shown in Fig.3(b).The original ESA at short time delays and the absence of anisotropy demonstrate that the ligand hole in the LMCT electronic excited state hops very quickly from ligand to ligand,making the excited state look delocalized on the vibrational time scale.This observation also implies that the measurement lacks su fficient temporal resolution to observe the initial dephasing and intra-ligand charge transfer rates represented by γ and Γ in Eq.(3).The eventual appearance of two distinct vibrational transitions with the same rise and decay time constants suggests a reduction in molecular symmetry associated with a localized excited state. As shown in Fig.3,the loss in symmetry causes two CN-stretch absorption peaks split by 20 cm−1.To experimentally resolve this 20 cm−1shift,a time resolution greater than 1 ps is needed.Otherwise,the two transitions will motionally narrow into a single transition that makes the vibrational spectroscopy insensitive to the ligand hole localization.This sub-picosecond loss of anisotropy found for degenerate electronic ex-cited states agrees with the tetraphenyl porphyrin results measured by Hochstrasser and co-workers[89]and ruthenium-tris-bipyridine results obtained by Hammerstr¨om and coworkers[88].But it is hard to apply directly the TRIR anisotropy measurements to ruthenium tris-bipyridine since there are no vibrational transitions in this molecule that can be easily mapped onto the charge transfer coordinates.Vibrational labeling with local vibrational modes,such as cyano groups,may help to solve this problem[91].
IV.PHOTO-INDUCED BOND ISOMERIZATION DYNAMICS
Photoisomerization process depends sensitively on the reaction environment.The di ff erences between liquid and protein solvated chromophores represent the most striking demonstration of photochemical sensitivity to local environment.In bacteriorhodopsin,retinal isomerizes around the C13=C14 double bond with a quantum yield of 0.6[92,93],while retinal isomerizes around multiple double bonds with signi fi cantly lower quantum yield in solution[94,95].As we discussed above,the photochemistry of HBDI chromophore also strongly depends on its surrounding environment[48−51].We believe that the detailed understanding of the relationship between reaction environment and the photochemical outcome has wideranging application including designing and directing light-driven materials and molecular sensors.Here,we show an example that uses TRIR anisotropy measurement to characterize the isomerization dynamics of julolidine malononitrile(JDMN),as illustrated in Fig.4,dissolved in dimethylsulfoxide(DMSO).Photoisomerization of a similar molecular system has been extensively studied which include the stilbene bond isomerization [96]and the twisted intramolecular charge transfer (TICT)proposed for the dual fl uorescence of 4-(N,N-dimethylamino)-benzonitrile(DMABN)[97].Previous investigations demonstrated the sensitivity of the photochemical dynamics to the details of the reaction environment,including the viscoelastic e ff ects and solvent electrostatic e ff ects[98−103].But a detailed understanding of the excited state isomerization dynamics in response to the environmental properties is still lacking.
As we mentioned above,the TRIR anisotropy can provide the relative angle between the electronic and vibrational transition dipole moments when molecular rotation can be ignored[38−42].As shown in Fig.5, photo-induced bond isomerization will change the relative angle θ.We place the rotational axisˆR along the z-axis since it is invariant in the molecular frame during the photoisomerization process.The relative angles θ between the electronic transition dipole momentµeand the vibrational transition dipole momentµvcan be correlated to the molecular structure through the following relationship:cosθ=cosθecosθv−sinθesinθvcosϕ, where ϕ=ϕe−ϕvis the dihedral angle between theˆRµeand theˆRµvplanes.So one can determine the bond rotation angle,ϕ,by measuring the angle θ and calculating the two tilt angles,θeand θv,from the combined experimental and computational studies.

FIG.4Molecular structures of julolidine malononitrile (JDMN).The electronic transition dipole and the torsional angles potentially involved in the electronic excited state relaxation dynamics are also shown.These fi gures are adapted with permission from Zhang et al.[23],copyright(2012) American Chemical Society.

FIG.5 Schematic view of how the photo-induced change in the torsional angle∆ϕ can be extracted from the polarization dependent TRIR spectroscopy.This fi gure is adapted with permission from Zhang et al.[23],copyright(2012) American Chemical Society.
The detailed assignment and analysis of photoinduced dynamics of JDMN in DMSO can be found in Ref.[23].In short,we have modeled the electronic excited state decay kinetics with two parallel relaxation channels each involving two sequential relaxation steps [23].Isotropic dynamics fi ts at three di ff erent central frequencies:2210 cm−1(GSB),2155 cm−1(ESA),and 2115 cm−1(ESA)with this kinetic model can be found in Fig.6(a).Figure 6(b)shows the time-dependent anisotropy measured at these three frequencies.The GSB has an initial anisotropy of 0.31±0.04 that does not have any decay in the 50 ps time window demonstrates that JDMN molecule rotation occurs on a time scale much slower than 50 ps,which suggests that the anisotropy values at long time delay can be used to as-sess the structural dynamics of the long-lived excited state.

FIG.6(a)Time-dependent change in transmission for the isotropic pump-probe signal.Population dynamics for the GSB measured at 2210 cm−1(∗),the ESA band measured at 2155 cm−1(□),and the ESA band measured at 2115 cm−1(◦)of JDMN measured in DMSO.(b)Timedependent anisotropy for JDMN in DMSO at three different spectra range:2210 cm−1(*),2155 cm−1(□)and 2115 cm−1(◦).These fi gures are adapted with permission from Zhang et al.[23],copyright(2012)American Chemical Society.
The ESA anisotropy measured at 2115 cm−1shows an initial value of 0.04±0.03 with no measurable time dependence,indicating the two electronic excited states have very similar anti-symmetric CN stretch anisotropies.However,the ESA anisotropy measured at 2155 cm−1has an initial value of 0.39±0.05 decaying to 0.13±0.03 which corresponds to a weighted sum of the two anisotropies[23].If both anisotropies were time independent,the time constant of the overall anisotropy would follow the 12.3 ps excited state population decay time constant.But this anisotropy decay occurs much faster than 12.3 ps strongly indicates that the anisotropy of long-lived excited state has a time-dependent decay.We have fi t it to a single exponential with an o ff set,r=Aexp(−t/τ)+C,which gives τ=2.6±0.7 ps,A=0.21±0.04,and C=0.18±0.03.
As shown in Fig.5,to correlate the measured relative transient dipole angle to the bond rotation,we need to calculate the tilt angles,θeand θv.TDDFT/CAMB3LYP calculations predict the presence of one structure minimum on the excited state potential energy surface(PES)near the Frank-Condon region that is consistent with no bond isomerization.The calculated PES energy varies weakly with torsion of bond b and bond c that is in line with the orientational dynamics observed for the excited state.A conical intersection is reached when bond c twists to τc≈80◦−90◦[98,104].We then searched the excited state PES and identi fi ed two di ff erent minima using TDDFT calculations.One minimum occurs in the Frank-Condon region and cannot account for the long-lived excited state that we labeled as an S1state.The second minimum occurs at τb=80◦that we labeled as Sbstate and corresponds to a rotational angle∆ϕ=80◦for bond b rotation[23].The Sbelectronic dipole moment exceeds that of S1by 6.2 Debye,which originates from the transfer of charge from the julolidine to the malononitrile π electron system.So the Sbstate can be assigned to a TICT state since it involves electron transfer between decoupled molecular orbitals [23,97].In short,we demonstrate that the combination of measurements and TDDFT calculations has confi rmed the photoisomerization of JDMN generates a metastable TICT excited state.We have to emphasize that there are two critical attributes to determine the success of this experiment.(i)To follow the structural dynamics using anisotropy measurement,it is necessary that the bond rotation changes the projection of the vibrational transition dipole moment onto the electronic transition dipole moment.(ii)The structural change in the laboratory frame and molecular frame needs to be similar.This requirement can be satis fi ed when the isomerizing bond separates the molecule into two components with very di ff erent moments of inertia,which is met by JDMN,but not by stilbene or azobenzene.V.FUTURE PROMISE
TRIR spectroscopy is particularly valuable in the study of systems containing CO or CN vibration mode since their frequencies and bandwidths sensitivities to electronic and molecular structure are well-established. But their extension to the larger biological system is still facing a lot of challenges,the major limitations being their low sensitivity and site-selectivity[105].The low site-selectivity is the consequence of the delocalized backbone vibrational mode and the possible frequent overlap with bu ff er solution.To improve the site-selectivity,researchers have developed various extrinsic vibrational probes and incorporated them into biological molecules to study their site-speci fi cal structural and environmental properties.These extrinsic vibrational probes have played an essential role in the study of a wide variety of structure and dynamics of proteins and peptides and the advancements in this rapidly growing research area have been inten-sively reviewed recently[106−108].The low sensitivity of the TRIR spectroscopy is the result of the low cross sections of vibrational transitions which are more than two orders of magnitude lower than those of the electronic transitions.Combining this low sensitivity with the strong absorption of water in the mid-IR region,biological samples need to be highly concentrated,which is not favorable for most of the larger proteins.However,there is a signi fi cant sensitivity gain when we move to the two-dimensional infrared(2DIR)spectroscopy.First,the detection can be background free when using the so-called box-CARS geometry.Second,the 2D-IR signal strength quadratically depends on the extinction coe ffi cient[105].For example,for a medium strong IR absorber with an extinction coe ffi cient of 500(mol/L)−1cm−1at 2120 cm−1in a cuvette of 10-micron thickness,an absorption band with 0.5 mOD is expected at 1 mmol/L concentration.The D2O background at the same condition is expected to be 100 mOD since the extinction coe ffi cient is 1.8(mol/L)−1cm−1at 2120 cm−1for D2O.So the signal ratio between the IR label and water background is 1:500 for linear spectroscopy,which is expected to be improved to 1:2.3 for 2D-IR measurement since the reason of the enormous water background is its massive concentration(~56 mol/L)[105].Combined with the development of other light-induced triggering,we expect the time-resolved 2D-IR spectroscopy will serve as a critical tool in providing new mechanistic insights into photo-induced biological problems[109,110].
Recently,high-intensity continuum mid-IR pulse has been demonstrated and used to study water hydrogen bonding related phenomenon[111−114].This advancement makes the TRIR more similar to the transient absorption experiment where the supercontinuum is commonly used.The extinction ratio of commercial mid-IR polarizer has also been dramatically improved which allows the researchers to detect extremely weak chiral signals on top of large achiral background contributions[115,116].The advancement of the 2D area detector has opened the opportunity to study the nonlinear infrared imaging of the heterogeneous samples with micron resolution[117].With all these technology and methodology developments,the structural dynamics study in the chemical and biological system using the ultrafast infrared methods will be furthered.
VI.ACKNOWLEDGMENTS
This work was supported by the“Thousand Plan”Youth Program and Beijing Normal University.
[1]S.Woutersen,U.Emmerichs,and H.J.Bakker,Science 278,658(1997)
[2]A.H.Zewail,J.Phys.Chem.A 104,5660(2000).
[3]N.A.Anderson and T.Q.Lian,Annual Rev.Phys. Chem.Palo Alto,491(2005).
[4]A.Rosspeintner,B.Lang,and E.Vauthey,Annual Review of Physical Chemistry,Vol.64,M.A.Johnson and T.J.Martinez,Eds.,Palo Alto:Annual Reviews, 247(2013).
[5]R.J.D.Miller,Science 343,1108(2014).
[6]J.R.Lakowicz,Principles of Fluorescence Spectroscopy,Springer,(2006).
[7]P.Mukherjee,Ultrafast Fluorescence Spectroscopy Used as a Probe to Explore Excited State Photophysics of Biologically and Environmentally Relevant Systems, Proquest:Umi Dissertation Publishing,(2011).
[8]Y.X.Weng and H.L.Chen,Ultrafast Spectroscopyrinciples and Techniques(Chinese Edition),Beijing: Chemical Industry Press,(2013).
[9]G.R.Fleming,Chemical ApplicationsofUltrafast Spectroscopy,Oxford:Oxford University Press, (1986).
[10]P.Hannaford,FemtosecondLaserSpectroscopy, Springer,(2005).
[11]M.D.Fayer,Ultrafast Infrared Vibrational Spectroscopy,New York:CRC Press,(2013).
[12]F.Schotte,H.S.Cho,V.R.I.Kaila,H.Kamikubo,N. Dashdorj,E.R.Henry,T.J.Graber,R.Henning,M. Wul ff,G.Hummer,M.Kataoka,and P.A.An fi nrud, Proc.Nat.Acad.Sci.USA 109,19256(2012).
[13]Y.O.Jung,J.H.Lee,J.Kim,M.Schmidt,K.Mo ff at, V.Srajer,and H.Ihee,Nature Chem.5,212(2013).
[14]K.H.Kim,J.G.Kim,S.Nozawa,T.Sato,K.Y. Oang,T.Kim,H.Ki,J.Jo,S.Park,C.Song,T. Sato,K.Ogawa,T.Togashi,K.Tono,M.Yabashi, T.Ishikawa,J.Kim,R.Ryoo,J.Kim,and H.Ihee, Nature 518(2015).
[15]H.T.Lemke,C.Bressler,L.X.Chen,D.M.Fritz,K. J.Ga ff ney,A.Galler,W.Gawelda,K.Haldrup,R.W. Hartsock,H.Ihee,J.Kim,K.H.Kim,J.H.Lee,M. M.Nielsen,A.B.Stickrath,W.Zhang,D.Zhu,and M.Cammarata,J.Phys.Chem.A 117,735(2013).
[16]W.Zhang,R.Alonso-Mori,U.Bergmann,C.Bressler, M.Chollet,A.Galler,W.Gawelda,R.G.Hadt,R. W.Hartsock,T.Kroll,K.S.Kjaer,K.Kubicek,H. T.Lemke,H.W.Liang,D.A.Meyer,M.M.Nielsen, C.Purser,J.S.Robinson,E.I.Solomon,Z.Sun,D. Sokaras,T.B.van Driel,G.Vanko,T.C.Weng,D. Zhu,and K.J.Ga ff ney,Nature 509,345(2015).
[17]P.Wernet,K.Kunnus,I.Josefsson,I.Rajkovic,W. Quevedo,M.Beye,S.Schreck,S.Grubel,M.Scholz, D.Nordlund,W.Zhang,R.W.Hartsock,W.F. Schlotter,J.J.Turner,B.Kennedy,F.Hennies,F. M.F.de Groot,K.J.Ga ff ney,S.Techert,M.Odelius, and A.Fohlisch,Nature 520,78(2015).
[18]W.Zhang and K.J.Ga ff ney,Account.Chem.Res.48, 1140(2015).
[19]B.J.Siwick,J.R.Dwyer,R.E.Jordan,and R.J.D. Miller,Science 302,1382(2003).
[20]A.H.Zewail,Annual Review of Physical Chemistry. Palo Alto:Annual Reviews,65(2006).
[21]M.B.Ji,M.Odelius,and K.J.Ga ff ney,Science 328, 1003(2010).
[22]D.Y.Vorobyev,C.H.Kuo,J.X.Chen,D.G.Kuroda, J.N.Scott,J.M.Vanderkooi,and R.M.Hochstrasser,J.Phys.Chem.B 113,15382(2009).
[23]W.K.Zhang,Z.G.Lan,Z.Sun,and K.J.Ga ff ney, J.Phys.Chem.B 116,11527(2012).
[24]D.McMorrow and W.T.Lotshaw,J.Phys.Chem.95, 10395(1991).
[25]A.M.Weiner,D.E.Leaird,G.P.Wiederrecht,and K.A.Nelson,J.Opt.Soc.Am.B 8,1264(1991).
[26]H.Hamaguchi,An.Rev.Phys.Chem.45,593(1994). [27]L.Dhar,J.A.Rogers,and K.A.Nelson,Chem.Rev. 94,157(1994).
[28]M.Schmitt,G.Knopp,A.Materny,and W.Kiefer, Chem.Phys.Lett.270,9(1997).
[29]P.Kukura,D.W.McCamant,and R.A.Mathies,Annual Review of Physical Chemistry,Palo Alto:Annual Reviews,461(2007).
[30]J.R.Schoonover and G.F.Strouse,Chem.Rev.98, 1335(1998).
[31]M.W.George and J.J.Turner,Coordination Chem. Rev.177,201(1998).
[32]E.T.J.Nibbering,H.Fidder,E.Pines,Annual Review of Physical Chemistry,Palo Alto:Annual Reviews, 337(2005).
[33]J.M.Butler,M.W.George,J.R.Schoonover,D.M. Dattelbaum,and T.J.Meyer,Coordination Chem. Rev.251,492(2007).
[34]R.D.Pensack,K.M.Banyas,L.W.Barbour,M. Hegadorn,and J.B.Asbury,Phys.Chem.Chem. Phys.11,2575(2009).
[35]H.J.Bakker and J.L.Skinner,Chem.Rev.110,1498 (2010).
[36]P.Hamm,Chimia 65,313(2011).
[37])M.D.Fayer and N.E.Levinger,Annual Review of Analytical Chemistry,Vol.3.E.S.Yeung and R.N. Zare Eds.,Palo Alto:Annual Reviews,89(2010).
[38]J.N.Moore,P.A.Hansen,and R.M.Hochstrasser, Proc.Nat.Acad.Sci.USA 85 5062(1988).
[39]M.Lim,T.A.Jackson,and P.A.An fi nrud,Science 269,962(1995).
[40]P.A.Hansen,J.N.Moore,and R.M.Hochstrasser, Chem.Phys.131,49(1989).
[41]M.H.Lim,T.A.Jackson,and P.A.An fi nrud,J. Chem.Phys.102,4355(1995).
[42]M.H.Lim,T.A.Jackson,and P.A.An fi nrud,Nature Struct.Bio.4,209(1997).
[43]P.A.An fi nrud,C.Han,and R.M.Hochstrasse,Proc. Nat.Acad.Sci.USA 86,8387(1989).
[44]D.E.Sagnella,J.E.Straub,T.A.Jackson,M.Lim, and P.A.An fi nrud,Proce.Nat.Acad.Sci.USA 96, 14324(1999).
[45]M.H.Lim,Bull.Korean Chem.Soc.23,865(2002). [46]T.Zemojtel,M.Rini,K.Heyne,T.Dandekar,E.T.J. Nibbering,and P.M.Kozlowski,J.Am.Chem.Soc. 126,1930(2004).
[47]H.Niwa,S.Inouye,T.Hirano,T.Matsuno,S.Kojima, M.Kubota,M.Ohashi,and F.I.Tsuji,Proc.Nat. Aca.Sci.USA 93,13617(1996).
[48]K.B.Bravaya,B.L.Grigorenko,A.V.Nemukhin, and A.I.Krylov,Acc.Chem.Res.45,265(2012).
[49]L.M.Tolbert,A.Baldridge,J.Kowalik,and K.M. Solntsev,Acc.Chem.Res.45,171(2012).
[50]R.Y.Tsien,Annual Rev.Biochem.67,509(1998).
[51]R.Y.Tsien,Angew.Chem.Int.Ed.48,5612(2009). [52]J.J.van Thor,K.L.Ronayne,M.Towrie,and J.T. Sage,Biophys.J.95,1902(2008).
[53]J.J.van Thor,Chem.Soc.Rev.38,2935(2009).
[54]A.M.Virshup,C.Punwong,T.V.Pogorelov,B.A. Lindquist,C.Ko,and T.J.Martinez,J.Phys.Chem. B 113,3280(2009).
[55]A.Usman,O.F.Mohammed,E.T.J.Nibbering,J. Dong,K.M.Solntsev,and L.M.Tolbert,J.Am. Chem.Soc.127,11214(2005).
[56]K.Heyne,O.F.Mohammed,A.Usman,J.Dreyer, and E.T.J.Nibbering,J.Am.Chem.Soc.127,18100 (2005).
[57]A.Usman,O.F.Mohammed,K.Heyne,J.Dreyer, and E.T.J.Nibbering,Chem.Phys.Lett.401,157 (2005).
[58]Y.Yang,M.Linke,T.von Haimberger,J.Hahn,R. Matute,L.Gonzalez,P.Schmieder,and K.Heyne,J. Am.Chem.Soc.134,1408(2012).
[59]S.K.Jha,M.B.A.Ji,K.J.Ga ff ney,and S.G.Boxer, Proc.Nat.Acad.Sci.USA 108,16612(2011).
[60]M.Linke,A.Lauer,T.von Haimberger,A.Zacarias, and K.Heyn,J.Am.Chem.Soc.130,14904(2008). [61]M.Linke,M.Theisen,T.von Haimberger,M.E. A.Madjet,A.Zacarias,H.Fidder,and K.Heyne, ChemPhysChem 11,1283(2010).
[62]M.Theisen,M.Linke,M.Kerbs,H.Fidder,M.E.A. Madjet,A.Zacarias,and K.Heyne,J.Chem.Phys. 131,8(2009).
[63]C.F.Wang,B.K.Mohney,B.B.Akhremitchev,and G.C.Walker,J.Phys.Chem.A 104,4314(2000).
[64]I.V.Rubtsov,N.P.Redmore,R.M.Hochstrasser,and M.J.Therien,J.Am.Chem.Soc.126,2684(2004). [65]R.A.Kaindl,M.Wurm,K.Reimann,P.Hamm,A. M.Weiner,and M.Woerner,J.Opt.Soc.Am.B 17, 2086(2000).
[66]K.Wynne and R.M.Hochstrasser,Chem.Phys.193, 211(1995).
[67]P.Hamm,Chem.Phys.200,415(1995).
[68]M.Chachisvilis,H.Fidder,and V.Sundstrom,Chem. Phys.Lett.234,141(1995).
[69]W.K.Zhang,M.B.Ji,Z.Sun,and K.J.Ga ff ney,J. Am.Chem.Soc.134,2581(2012).
[70]R.Jimenez,S.N.Dikshit,S.E.Bradforth,and G.R. Fleming,J.Phys.Chem.100,6825(1996).
[71]C.Sissa,A.Painelli,M.Blanchard-Desce,and F. Terenziani,J.Phys.Chem.B 115,7009(2011).
[72]D.M.Jonas,M.J.Lang,Y.Nagasawa,T.Joo,and G.R.Fleming,J.Phys.Chem.100,12660(1996).
[73]K.Wynne,S.M.Lecours,C.Galli,M.J.Therien, and R.M.Hochstrasser,J.Am.Chem.Soc.117,3749 (1995).
[74]R.Kumble,S.Palese,V.S.Y.Lin,M.J.Therien,and R.M.Hochstrasser,J.Am.Chem.Soc.120,11489 (1998).
[75]C.K.Min,T.Joo,M.C.Yoon,C.M.Kim,Y. N.Hwang,D.Kim,N.Aratani,N.Yoshida,and A. Osuka,J.Chem.Phys.114,6750(2001).
[76]W.Qian and D.M.Jonas,J.Chem.Phys.119,1611 (2003).
[77]D.A.Farrow,E.R.Smith,W.Qian,and D.M.Jonas, J.Chem.Phys.129,20(2008).
[78]O.Schalk and A.N.Unterreiner,Phys.Chem.Chem. Phys.12,655(2010).
[79]E.R.Smith and D.M.Jonas,J.Phys.Chem.A 115,4101(2011).
[80]A.J.Van Tassle,M.A.Prantil,and G.R.Fleming, J.Phys.Chem.B 110,18989(2006).
[81]J.Rehault,V.Zanirato,M.Olivucci,and J.Helbing, J.Chem.Phy.134,10(2011).
[82]G.S.Engel,T.R.Calhoun,E.L.Read,T.K.Ahn, T.Mancal,Y.C.Cheng,R.E.Blankenship,and G. R.Fleming,Nature 446,782(2007).
[83]E.Collini and G.D.Scholes,Science 323,369(2009). [84]R.S.Knox and D.Gulen,Photochem.Photobiol. 57,40(1993).
[85]K.Wynne and R.M.Hochstrasser,J.Raman Spectro. 26,561(1995).
[86]R.A.Malone and D.F.Kelley,J.Chem.Phys.95, 8970(1991).
[87]A.T.Yeh,C.V.Shank,and J.K.McCusker,Science 289,935(2000).
[88]S.Wallin,J.Davidsson,J.Modin,and L.Hammarstrom,J.Phys.Chem.A 109,4697(2005).
[89]C.Galli,K.Wynne,S.M.Lecours,M.J.Therien, and R.M.Hochstrasser,Chem.Phys.Lett.206,493 (1993).
[90]R.J.Sension,S.T.Repinec,A.Z.Szarka,and R.M. Hochstrasser,J.Chem.Phys.98,6291(1993).
[91]C.E.McCusker and J.K.McCusker,Inorg.Chem. 50,1656(2011).
[92]R.A.Mathies,S.W.Lin,J.B.Ames,and W.T.Pollard,Annual Rev.Biophys.Biophys.Chem.20,491 (1991).
[93]R.Neutze,E.Pebay-Peyroula,K.Edman,A.Royant, J.Navarro,and E.M.Landau,Biochim.Et Biophys. Acta-Biomembr.1565,144(2002).
[94]P.Hamm,M.Zurek,T.Roschinger,H.Patzelt,D. Oesterhelt,and W.Zinth,Chem.Phys.Lett.263,613 (1996).
[95]P.Hamm,M.Zurek,T.Roschinger,H.Patzelt,D.S. Oesterhelt,and W.Zinth,Chem.Phys.Lett.268,180 (1997).
[96]D.H.Waldeck,Chem.Rev.91,415(1991).
[97]Z.R.Grabowski,K.Rotkiewicz,and W.Rettig, Chem.Rev.103,3899(2003).
[98]C.Swalina and M.Maroncelli,J.Phys.Chem.C 114, 5602(2010).
[99]H.Jin,M.Liang,S.Arzhantsev,X.Li,and M.Maroncelli,J.Phys.Chem.B 114,7565(2010).
[100]B.D.Allen,A.C.Benniston,A.Harriman,S.A.Rostron,and C.F.Yu,Phys.Chem.Chem.Phys.7,3035 (2005).
[101]A.Y.Jee,E.Bae,and M.Lee,J.Phys.Chem.B 113, 16508(2009).
[102]K.I.Gutkowski,M.L.Japas,and P.F.Aramendia, Chem.Phys.Lett.426,329(2006).
[103]A Paul and A Samanta,J.Phys.Chem.B 112,16626 (2008).
[104]Z.G.Lan,Y.Lu,O.Weingart,and W.Thiel,J.Phys. Chem.A 116,1510(2012).
[105]K.L.Koziol,P.J.M.Johnson,B.Stucki-Buchli,S. A.Waldauer,and P.Hamm,Curr.Opin.Struct.Biol. 34,1(2015).
[106]M.M.Waegele,R.M.Culik,and F.Gai,J.Phys. Chem.Lett.2,2598(2011).
[107]H.Kim and M.Cho,Chem.Rev.113,5817(2013).
[108]J.Q.Ma,I.M.Pazos,W.K.Zhang,R.M.Culik,and F.Gai,Annual Review of Physical Chemistry,Vol 66, M.A.Johnson and T.J.Martinez,Eds.,Palo Alto: Annual Reviews,357(2015).
[109]M.J.Tucker,M.Abdo,J.R.Courter,J.X.Chen,S. P.Brown,A.B.Smith,and R.M.Hochstrasser,Proc. Natl.Acad.Sci.USA 110,17314(2013).
[110]A.A.Deeg,M.S.Rampp,A.Popp,B.M.Pilles,T. E.Schrader,L.Moroder,K.Hauser,and W.Zinth, Chem.Eur.J.20,694(2014).
[111]A.B.Myers and R.M.Hochstrasser,J.Chem.Phys. 85,6301(1986).
[112]P.B.Petersen and A Tokmako ff,Opt.Lett.35,1962 (2010).
[113]L.De Marco,M.Thamer,M.Reppert,and A.Tokmako ff,J.Chem.Phys.141,10(2014).
[114]M.Thamer,L.De Marco,K.Ramasesha,A.Mandal, and A.Tokmako ff,Science 350,78(2015).
[115]M.Bonmarin and J.Helbing,Chirality 21,E298 (2009).
[116]H.J.Rhee,Y.G.June,J.S.Lee,K.K.Lee,J.H Ha, Z.H.Kim,S.J.Jeon,and M.H.Cho,Nature 458, 310(2009).
[117]C.R.Baiz,D.Schach,and A.Tokmako ff,Opt.Express 22,18724(2014).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study†
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study†
- ARTICLE Infrared Photodisssociation Spectroscopy of Boron Carbonyl Cation Complexes†