
基于文本挖掘的高通量癌症基因组数据注释
2016-03-22 03:57,,,,
中华医学图书情报杂志 2016年12期
,, , ,
目前,癌症已成为威胁人类健康最危险的杀手之一。中国癌症统计报告指出,2015年中国有429.2万例新发肿瘤病例和281.4万例死亡病例,且发病率和死亡率还在不断上升,癌症已经成为最重要的健康问题之一[1],其相关领域的研究也受到了越来越广泛的关注。癌症和肿瘤基因图谱(The Cancer Genome Atlas,TCGA)计划的提出加速了高通量癌症基因组数据资源的产生,面对大量非结构化的数据资源,如何从中获取有价值的信息和潜在的关系变得至关重要,从大规模自由文本中自动获取、挖掘有意义的知识是一项迫切的任务。虽然目前已经有很多文本挖掘、命名实体识别的技术和工具,自动识别也不再是问题,但这些工具和方法各有利弊,并不能完全满足实际需要。
本文主要围绕癌症基因组数据资源、高通量癌症基因组数据挖掘以及特异癌症基因描述文本提取与命名实体识别展开。
1 研究对象
癌症的发生是由基因变异导致的,癌症基因组数据的分析和利用成为解决癌症问题的基础[2]。癌症基因组学计划是一个公共项目,旨在发现全面的癌症基因组测序数据集,以帮助提高癌症诊断方法和治疗标准,并最终达到预防癌症的目的[3]。目前,基于已有的数据集,研究者做了大量的分析和挖掘工作[4-7]。另外,为了帮助科研人员从高通量癌症基因组数据中获取有价值的信息,开展了大量癌症基因组数据挖掘工具的研发。搜索引擎Zodiac利用贝叶斯图形模型与似然模型相结合的分析方法,通过对高通量TCGA癌症基因组数据的挖掘研究,从而返回特异基因及其拷贝数、基因表达、甲基化、蛋白质表达等信息[8]。目前,Zodiac已包含大约2亿个基因网络,所有相互作用网络的统计推理都是基于癌症基因组数据,它可以帮助我们了解基因在肿瘤疾病中的作用及基因之间的相互作用等,进而发现潜在的药物靶标、遗传畸变等。
面对从高通量癌症基因组学数据挖掘出来的特异基因和基因变异,需要借助文本描述和注释,以帮助肿瘤研究人员解读挖掘结果[9]。借助文本挖掘技术和已有的生物医学主题词表、数据库等知识才能快速发现有价值的关键信息[10-11]。生物医学文本挖掘是从无结构化文本中定位出具有特定语义类型的片段(如疾病、基因、药物命名实体识别),并且识别特定语义关系(如疾病的诊断基因标志物),生物医学文本挖掘技术包括文本检索、命名实体识别、关系提取、文本摘要、问答系统等[12-13]。命名实体识别是指从文本中识别出专有名称和特定类型的实体,如疾病和化学药物的名称等,命名实体识别技术是信息抽取、机器翻译、问答系统等多种自然语言处理技术必不可少的组成部分,也是本文采用的主要技术手段[14-15]。
2 数据集
2006年,美国启动了TCGA计划,试图通过基因组分析技术,特别是采用大规模的基因组测序数据,将人类全部癌症的基因组变异图谱绘制出来,并进行系统分析,进而发现新的诊断和治疗方法。TCGA是以基因组为基础的研究计划,通过广泛的合作,了解与病理机制相关的基因组变化,以及恶性肿瘤形成、生长、转移等的生物学基础,加速对癌症早期诊断及治疗的步伐,有效防止癌症的发生[16]。本文通过对本质、特性以及基因之间的关联关系等功能描述信息进行研究,如发现有价值的潜在信息。
近年来,乳腺癌已经成为困扰女性健康的重要因素,乳腺癌的发病人数和死亡人数都在不断增加,引起了社会各界的广泛关注。因此,如何尽快找到与乳腺癌相关的基因,并从中发现诊断、治疗乳腺癌的方法非常迫切。Zodiac通过对高通量癌症基因组学数据的分析研究,发现了一组与癌症显著相关的基因,并从TCGA相关的高通量开放数据库中获取到一系列权威的基因功能描述文本,其中包括乳腺癌相关的基因,如BCAR3、BEX2等。面对大量非结构化的自由文本,通过系统的文本挖掘与注释方法才能快速、有效地挖掘出对癌症诊断和治疗有意义的文本片段。本文从TCGA的高通量数据集中共获取了11 821条癌症相关基因数据,包括基因名称(Gene Name)和对该基因的功能描述(Gene Summary)。以乳腺癌相关的两个基因BCAR3和BEX2为例,其基因功能描述文本分别如图1和图2所示,其中包含疾病名称(breast cancer、glioma)及相关的药物名称(estrogen、tamoxifen)、基因名称(gene3)和蛋白质名称(CDC48)等信息。
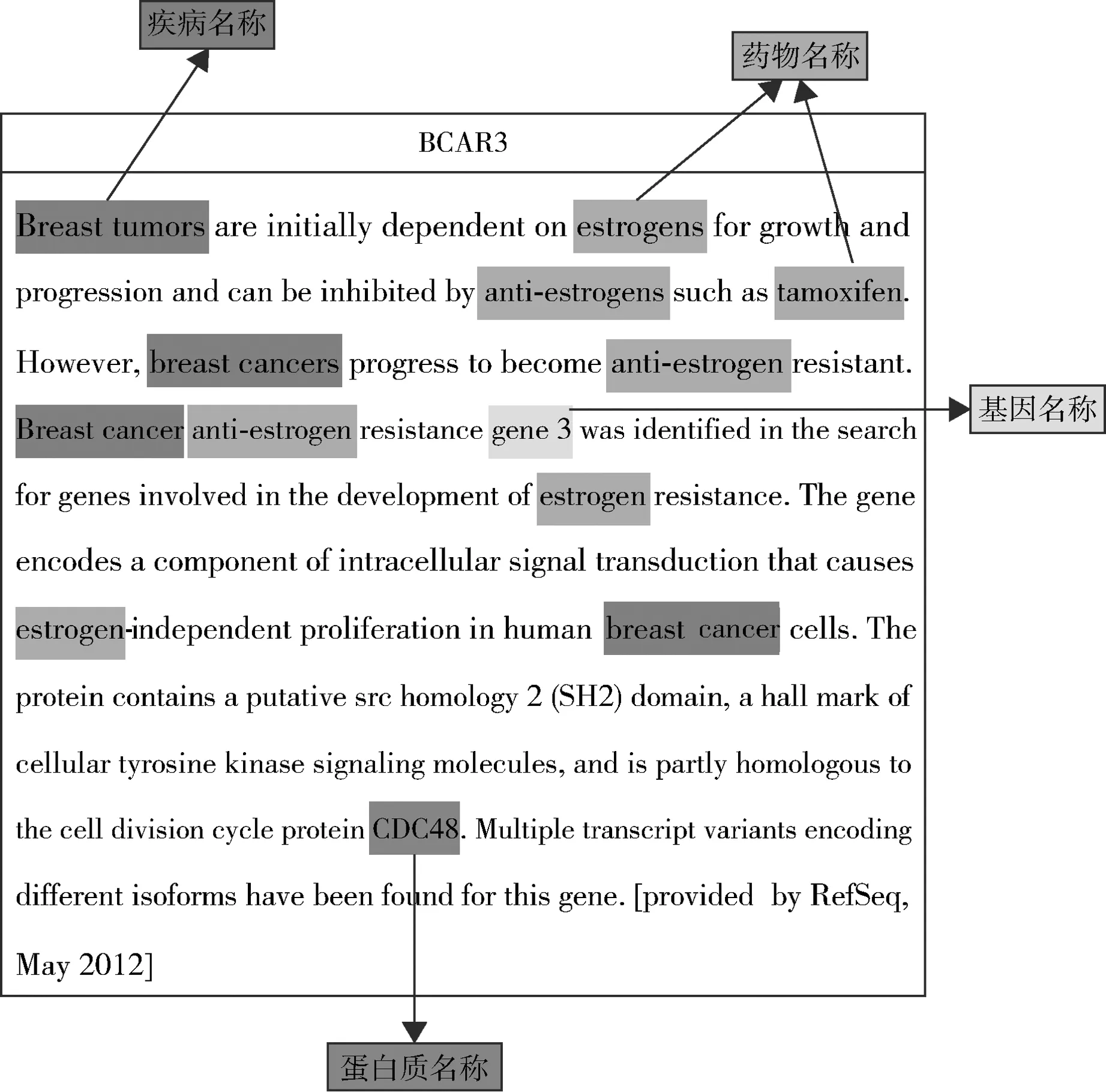
图1 BCAR3 - 乳腺癌相关基因数据样
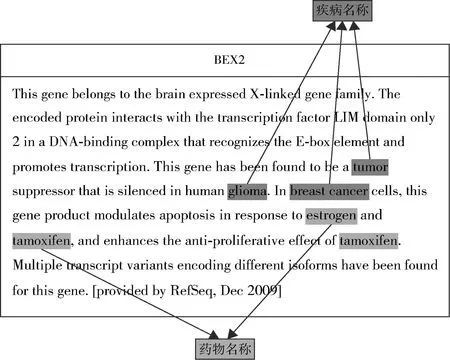
图2 BEX2 - 乳腺癌相关基因数据样
以上述基因功能描述文本为数据集,从中进行疾病、药物命名实体等的分析和挖掘,以帮助癌症研究者更好地发现诊断癌症的方法和治疗癌症的药物。
3 研究方法
本文基于上述高通量癌症基因组数据和生物医学领域科技词表、数据库等,应用生物医学领域文本挖掘、分析等方法,准确、高效地识别出关键的疾病和化学药物信息,经过规范化的分析处理来直观、可视化地展示结果,从而极大地方便相关研究人员发现有价值的疾病、药物以及基因之间的关系。基因功能描述文本数据的处理流程如图3所示。
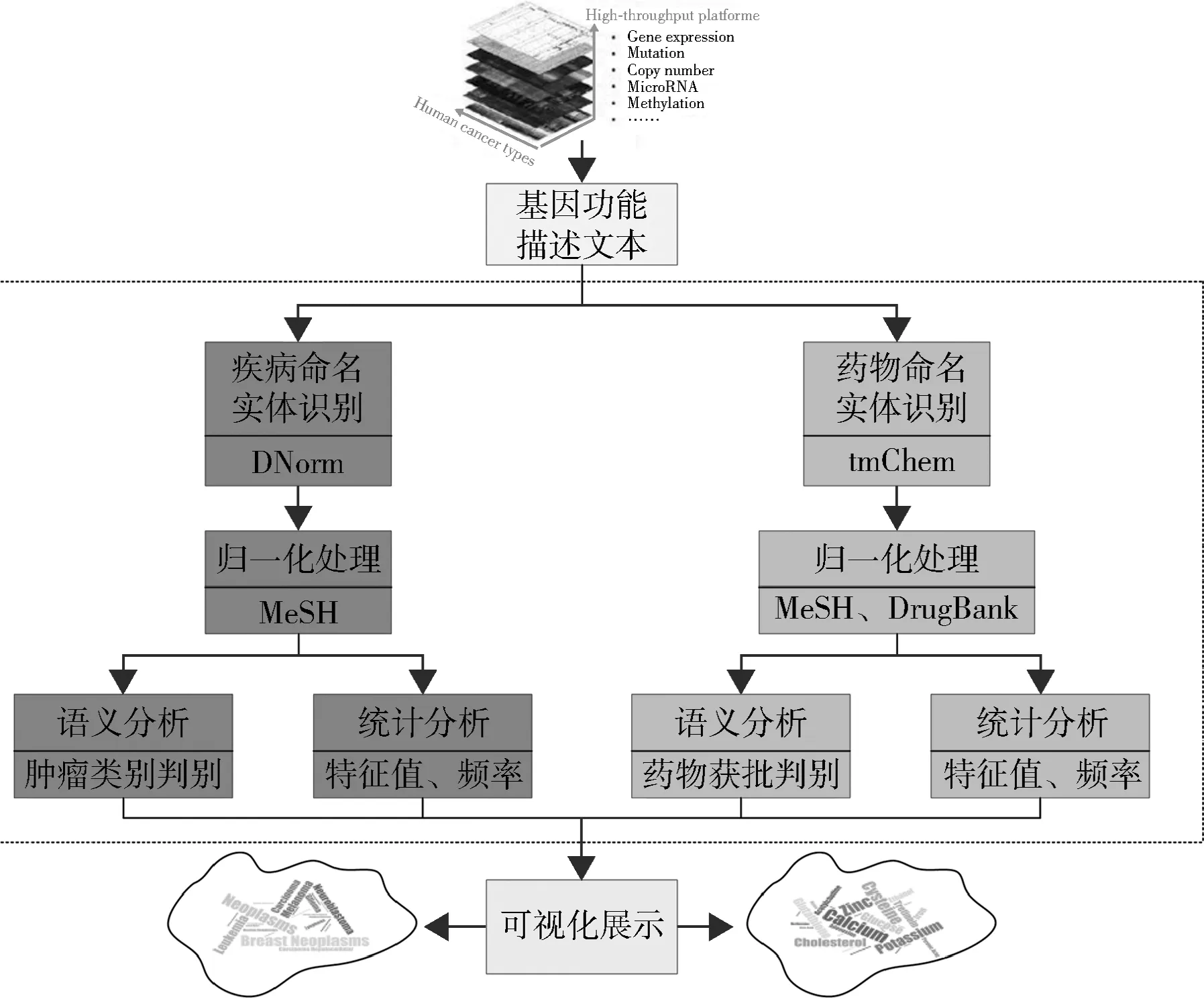
图3 基因功能描述文本的数据处理流程
首先,从海量TCGA相关的癌症基因组学数据中获取到一系列基因功能描述文本,并通过疾病、药物命名实体识别工具识别出有效的疾病、药物命名实体;其次,针对疾病和药物命名实体,分别通过规范化处理方法进行处理;第三,利用语义分析和多角度的统计分析方法分别得到语义分析结果(实体类别)和统计分析结果(特征值、频率)信息;最后,使用可视化的方法直观展示上述分析结果。
3.1 命名实体识别
目前,文本挖掘的开源工具有很多,但大多只适用于特定的应用场景,且一般会有其独特的格式要求,很难直接满足特定需求。本文应用生物医学命名实体识别技术识别自由文本中基因相关的疾病和药物[17-19],使用由美国国立医学图书馆(NCBI)开发的命名实体识别工具[20-22],针对疾病和化学药物的工具分别为DNorm-0.0.6和tmChemM1-0.0.2。以上述基因功能描述样本BCAR3和BEX2为例,分别介绍疾病和药物命名实体识别结果。
通过对上述癌症基因文本数据集进行疾病命名实体的识别,共识别出11 502个疾病实体。基因BCAR3和BEX2的疾病命名实体识别结果如图4所示,每一行代表识别出的一个疾病实体,包含的信息有基因名称(Official Symbol)、疾病在该基因文本中出现的起始位置(Start Position)和结束位置(End Position)、疾病术语(Disease Mention)、实体类型(Entity Type)和实体的概念ID(Concept ID)。
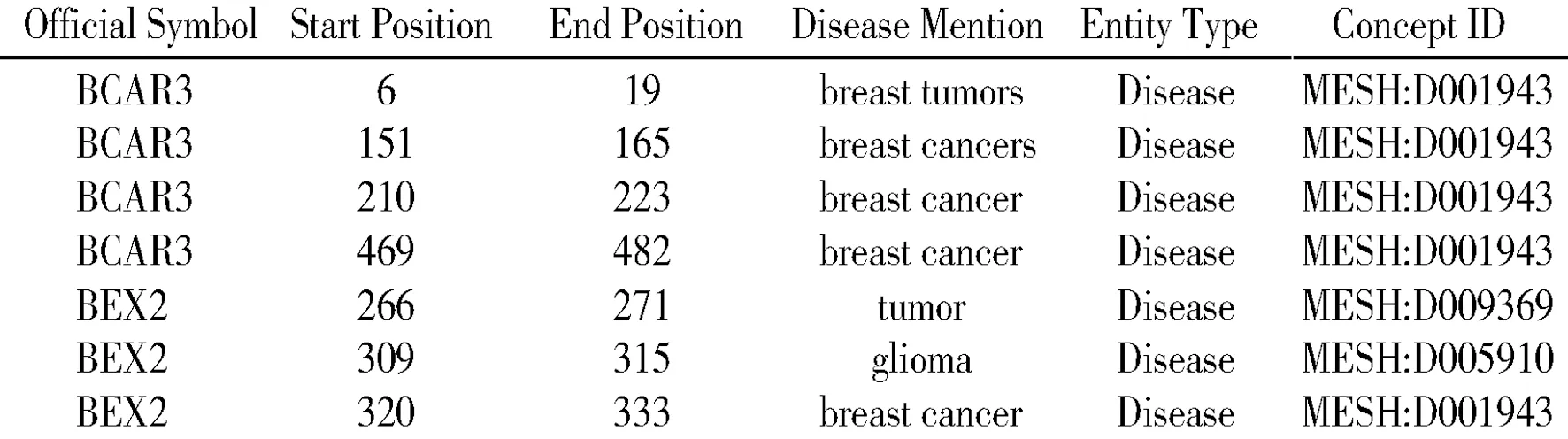
图4 BCAR3、BEX2疾病命名实体识别结果
通过对上述癌症基因文本数据集进行药物命名实体的识别,共识别出13 024个药物实体。图5表示基因BCAR3和BEX2的药物命名实体识别结果。
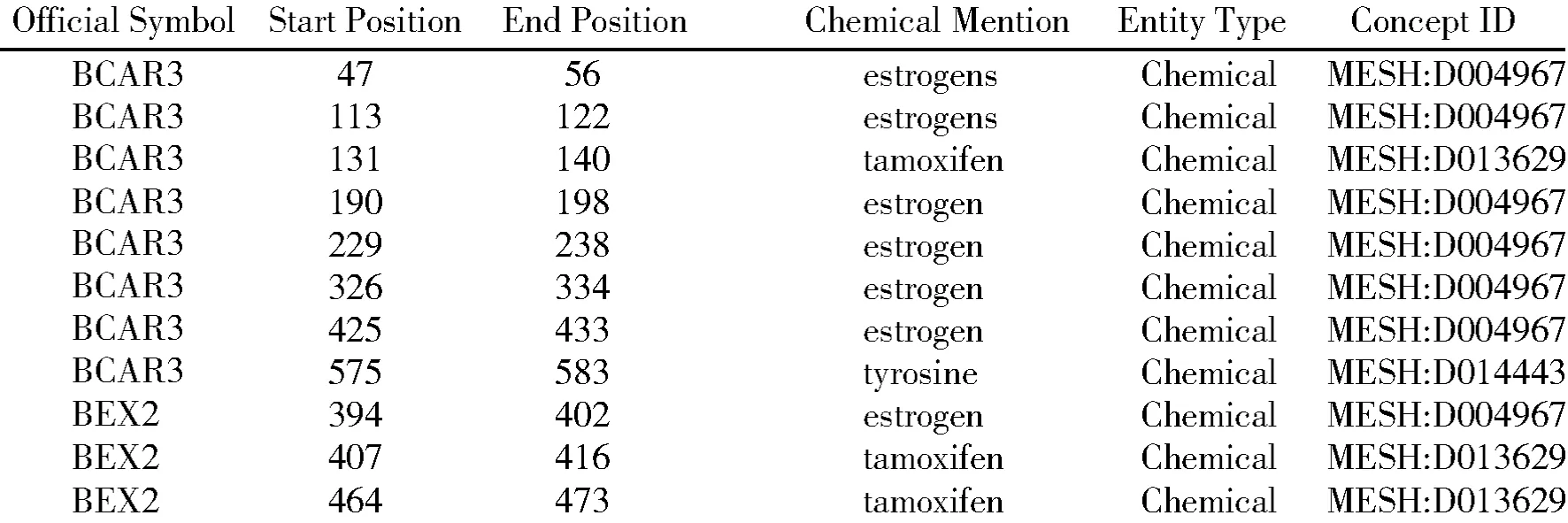
图5 BCAR3、BEX2药物命名实体识别结果
3.2 规范化处理
由上述命名实体识别工具识别出的结果中存在大量冗余信息,缺乏去噪、规范化等处理,也未对各术语出现的频率、关联关系等进行统计和分析,尚不能满足项目和后续研究的需要。因此,为了得到更准确、更有价值的疾病和药物命名实体识别结果,有必要通过一系列后处理工作来完善结果。
对于疾病命名实体识别,我们对从上述大量基因描述文本中识别出所有疾病相关的术语进行规范化处理,即映射至医学主题词表MeSH(Medical Subject Headings, https://www.nlm.nih.gov/mesh/ )。此外,我们还提供了相关疾病的特征信息,如术语的频率、该术语总共出现在几个基因描述文本中、术语是否与癌症相关等。
具体处理步骤如下:如果缩略术语在基因描述文本中有全名,则将概念ID对应的标准名称赋予该术语,以保证结果中同一概念ID对应的名称全部一致,且都为标准名称;如果在基因描述文本中找不到该缩略术语的全名,并且没有相应的概念ID信息或ID为-1,则删除该术语对应的结果;如果在基因描述文本中找不到该缩略术语的全名,但是中间结果中含有相应的概念ID信息,则降低该术语的频率;将所有概念ID为OMIM(Online Mendelian Inheritance in Man,http://omim.org/)的术语映射至MeSH词表,并根据CTD疾病词典将概念ID与全名进行映射。经过后处理,识别出的疾病术语均被映射到相应的概念名称(ConceptName)如图6所示,疾病术语“Breast tumors”“breast cancers”“Breast cancer”“breast cancer”都被映射至“Breast Neoplasms”,“tumor”和“glioma”分别被映射到标准名称“Neoplasms”和“Glioma”。

图6 疾病全名映射
对于药物命名实体识别,我们对从上述基因描述文本中识别出的所有药物相关的术语进行规范化处理映射至医学主题词表MeSH,同样提供了如术语频率、是否与药物相关等特征信息。具体处理步骤如下:如果缩略术语在基因描述文本中有全名,则将概念ID对应的标准名称赋予该术语,以保证结果中同一概念ID对应的名称全部一致,且都为标准名称;如果在基因描述文本中找不到该术语的全名,并且没有相应的概念ID信息或ID为-1,则删除该术语对应的结果;根据CTD药物词典,将概念ID与全名进行映射。经后处理,识别出的疾病术语均被映射到相应的概念名称(ConceptName)如图7所示,药物术语“estrogens”和“estrogen”被映射至“Estrogens”,“tamoxifen”和“tyrosine”分别被映射到标准名称“Tamoxifen”和“tyrosine”。

图7 药物全名映射
3.3 可视化
标签云(tag cloud)是一种展示关键词的新型可视化方法,可根据标签的权重大小区分其重要程度。一般来说权重越大的标签,在标签云中的字体就会越大、视觉效果会越好,也就会更容易被用户所关注。标签的可视化属性体现不同的权重程度,可以通过对云中标签可视化属性的操作对用户浏览产生一定的导向作用,把用户的关注点吸引到热门字段。本文使用了标签云生成工具对识别出的疾病和药物命名实体进行了可视化展示,并应用于Zodiac系统中,为用户提供更方便、更直观的体验。在上述命名实体识别工具和后处理的基础上,可根据不同实体出现的频次高低决定其重要与否,选择频次较高的部分实体作为标签内容,让用户能快速发现并了解热门标签——与基因相关的疾病和药物。
4 结果与分析
4.1 疾病命名实体识别
经过疾病命名实体识别和规范化处理分析,我们首先获取了每个基因功能描述文本中的所有疾病实体,并进一步得出了实体所属类别、是否与癌症相关、出现的次数及频率等,进而得到规范化的疾病命名实体识别结果。最终结果中包含的具体信息有概念名称(ConceptName)、概念ID(ConceptID)、基因名称(GeneName)、术语(Mention)、所属类别(Category)、是否癌症相关(IsCancerRelated)、基因次数(GeneCount)、出现频次(OverallFreq.)、词频(TF)。
其中,概念ID是从MeSH主题词表中获取的,并对同一ID所对应的不同形式术语进行规范化处理,得到统一的概念名称;基因名称是指疾病术语的出处所在;术语是实体在基因文本中的原始描述,含缩略词、同义词等,根据该实体的术语类型和所属类别判断是否与癌症相关,相关为“1”,不相关则为“0”;基因次数是计算该术语总共出现在了多少个基因文本中,可以在一定层面上说明该术语的重要程度;出现频次则为该术语在所有的基因文本中总出现的次数;词频的本义是指某一个词在某文件中出现的次数,是一种用于文本挖掘的常用技术,用以评估一个词语对于某一文本或文本集的重要程度,词频越高则越重要,本文中词频的计算则同时考虑了出现在某一文本中的所有实体的总数和包含该实体的文本个数这两个因素,先计算每个基因文本中某术语出现的次数除以该基因中识别出的术语总数,再将出现该术语的所有情况进行相加,得到该概念的词频。如公式1所示,其中n为包含该实体的文本个数,Nk为该术语在某一文本中出现的次数,Sk为该术语所在文本中识别出的实体总数。
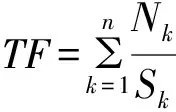
BCAR3和BEX2的处理结果如表1所示。其中,基因BCAR3 的功能描述文本中共识别出4个与癌症相关的实体,且均为乳腺癌,能够在一定程度上表明该基因与乳腺癌密切相关,进而方便癌症研究者从中发现诊断和治疗乳腺癌的有效方法。基因BEX2则包含三个不同的实体,且都与癌症相关,研究者可以从中探索三者之间是否存在潜在的关联关系,同样能够为防治癌症提供有力依据。另外,不同概念实体出现的频次、频率等结果也可能是研究者挖掘显著基因和实体的关键信息。

表1 规范化处理得到的疾病命名实体识别结果
4.2 药物命名实体识别结果
与疾病命名实体处理类似,我们得到的药物相关结果包括概念名称(ConceptName)、概念ID(ConceptID)、基因名称(GeneName)、术语(Mention)、术语类型(MentionType)、药物数据库ID(DrugBankID)、是否与药物相关(IsDrugRelated)、基因次数(GeneCount)、出现频次(OverallFreq.)、词频(TF)。其中,药物数据库ID是记录该术语在DrugBank中是否存在,存在则为“1”,不存在则为“-1”或“null”。表2为BCAR3和BEX2的药物处理结果。
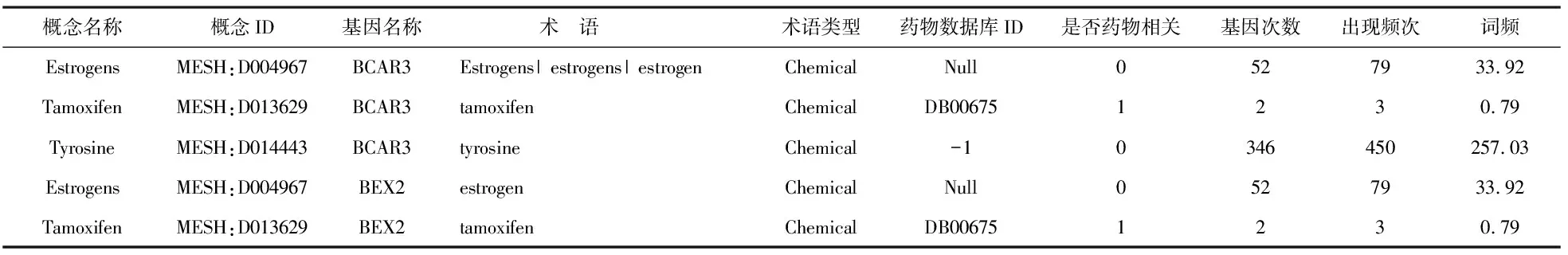
表2 规范化处理得到的药物命名实体识别结果
从表2可看出,基因BCAR3 的功能描述文本中共识别出3种化学命名实体,其中Tamoxifen被DrugBank药物数据库所收录,而Estrogens出现了7次。根据收录情况和出现的次数来看,二者都极有可能与乳腺癌的诊断和治疗有一定关联。从基因BEX2中共识别出2种化学命名实体,即Tamoxifen和Estrogens,研究者也可以从中挖掘二者是否与乳腺癌存在潜在的关联关系,为防治癌症提供有力的依据。事实上,研究者们已经证实了雌激素(Estrogens)在乳腺癌的发生和治疗中均起着至关重要的作用[23-24]。另外,不同概念实体出现的频次、频率等结果也可能是研究者挖掘显著基因和实体的关键信息。如tyrosine的“出现频次”“基因次数”“词频”相对较高,可能与乳腺癌存在着一定的关联关系。
5 结论和展望
本文围绕高通量癌症基因组学相关文本注释的方法、技术和应用等问题展开介绍,包括生物医学文本挖掘的相关技术,数据采集、命名实体识别、规范化处理、标签云等,并对疾病和药物命名实体识别的结果进行了规范化分析,同时针对乳腺癌相关的两个样例基因文本进行了全面分析。
目前,国内关于生物医学文本挖掘方面的研究相对欠缺,该领域的方法还不够成熟,没有统一的标准和工具,针对不同的应用场景必须进行相应的设计、调整。如对于英文生物医学文本的挖掘,已有大量的研究成果,但无法直接用于中文的分词、标注、识别等,亟待进一步地解决。接下来的研究中,我们将重点关注中文领域的生物医学文本挖掘技术及标准化较高的方法和工具,进一步提高生物医学领域与国际同行间研究的可比性。
猜你喜欢
今日农业(2021年11期)2021-08-13
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中成药(2018年7期)2018-08-04
东方女性(2018年3期)2018-04-16
散文诗(2017年17期)2018-01-31
中国科技术语(2012年5期)2012-03-20
中国科技术语(2012年3期)2012-03-20
中国科技术语(2012年3期)2012-03-20
