
桉树青枯病病原菌的分子鉴定及其致病性测定
2016-03-03 03:12:22谢耀坚李国清吴志华
桉树科技 2016年4期
王 艳,谢耀坚,李国清,吴志华
桉树青枯病病原菌的分子鉴定及其致病性测定
王艳1,2,谢耀坚1,李国清1,吴志华1*
(1.国家林业局桉树研究开发中心, 广东湛江 524022;2. APP中国林务事业部, 海南儋州578101;)
对华南4个地区的桉树青枯病病原菌进行了分子鉴定和致病性测试研究,以期为有效预防其发生提供依据。分别对来源于不同区域的11株桉树青枯病病原菌进行分子鉴定和致病力强弱测试比较,11株病原菌与序列形成支持率为86%分枝,表明其病原菌16S rRNA 基因序列与具有高度同源性。以桉树无性系DH32-29组培苗为材料,采用伤根法对11个菌株进行致病力测试,不同菌株间在致病性上差异显著(<0.01或<0.05),以发病高峰期、高峰期发病率和平均发病率进行K-均值聚类分析(K=3),结果表明来源于广东、广西、海南的各菌株间致病性存在差异,HY01、HY02、DA01、HP04菌株与HP05分为1类,DA02和DA03为第2类,而YJ01、HP01、HP02和HP03为第3类。对4个不同地区(采样点)的青枯病菌株平均发病率进行方差分析,结果发现来源广东阳江菌株致病性显著地低于其他来源菌株。不同地理来源菌株之间在致病性上无明显相关性,同一地区内同时存在着致病力强弱菌株,HP03的生物型4-1菌株与其他生物型3菌株在致病性亦无明显的差别。
桉树青枯病;分子鉴定;致病性;生物型
青枯病(Bacterial wilt)是由引起的土传性细菌病害[1]。近年来,我国南方桉树()、木麻黄()、桑树()等林木青枯病害的发生,造成较大经济损失,已成为林业上一种毁灭性病害[2]。桉树青枯病最早在我国广西柳桉()、巨桉()等幼树上首次发现[3]。广东、广西及海南是我国桉树人工林主产区,随着桉树人工林不断发展,桉树青枯病发生日趋严重,为害面积和分布范围逐年增大,对桉树人工林发展造成灾难性损失,已成为当前限制我国桉树发展的主要因子之一。
青枯病病原菌经寄主根的伤口或者次生根根冠部侵入,然后通过木质部繁殖扩散[4],形成侵填体[5],堵塞植物的维管束系统[6-7],造成寄主植物迅速萎蔫、枯死,而茎叶仍保持绿色[8]。目前生产上的桉树品种均不同程度上受其危害。特别是台风袭击后,感病品系幼林发病率达20% ~ 40 %,高感品系甚至60%以上[9]。桉树品种受到侵染后,将大幅度降低其生长势,影响桉树产量和林木质量,严重威胁着桉树人工林可持续发展。
尽管可通过培育抗病品种、改变不同耕作如轮作等措施防治其发生,但由于受立地土壤类型、病原菌种源、栽培模式、气候因素和品种等因素影响,加之青枯病病原菌的危害程度大、分布广、土壤存活时间长,青枯病仍然是桉树生产实际上面临的重要障碍。
吴清平等[10]发现广东雷州不同桉树树种分离获得的青枯菌菌株,其致病力基本相同,病原菌对桉树的致病力比对木麻黄、番茄()、甘薯()及花生()强。王胜坤[11]对采集于华南地区的30个桉树青枯菌菌株进行了致病性测试,结果表明青枯菌菌株的致病力和地理来源相关性不明显,地理来源相距较远的菌株,致病力差异不显著,而在同一地区,菌株致病力存在强弱分化,差异显著。由于桉树青枯病病原菌复杂,给生产和防治带来许多的困难和挑战。因此,本试验对来源于华南不同地区11株病原菌进行了分子鉴定和致病性测试等研究,以期为有效预防其发生和培育抗性品系提供依据。
1 材料和方法
1.1 材料
供试病原菌于2011年9―10月分别从华南3省各地不同桉树采集(表1),采集的病株均已表现出青枯病典型症状,即病株叶片失水萎蔫,伐倒后木质部呈黑褐色,片刻可见乳白色至淡黄褐色菌脓溢出。纯化后的菌株采用-70℃长期保存,或置无菌水中保存于室温或4℃下数月到数年,本实验保存于无菌水中。病原菌的分离活化培养采用CPG、TZC和NA等培养基。经前期形态学鉴定确定其病原菌为[12]。
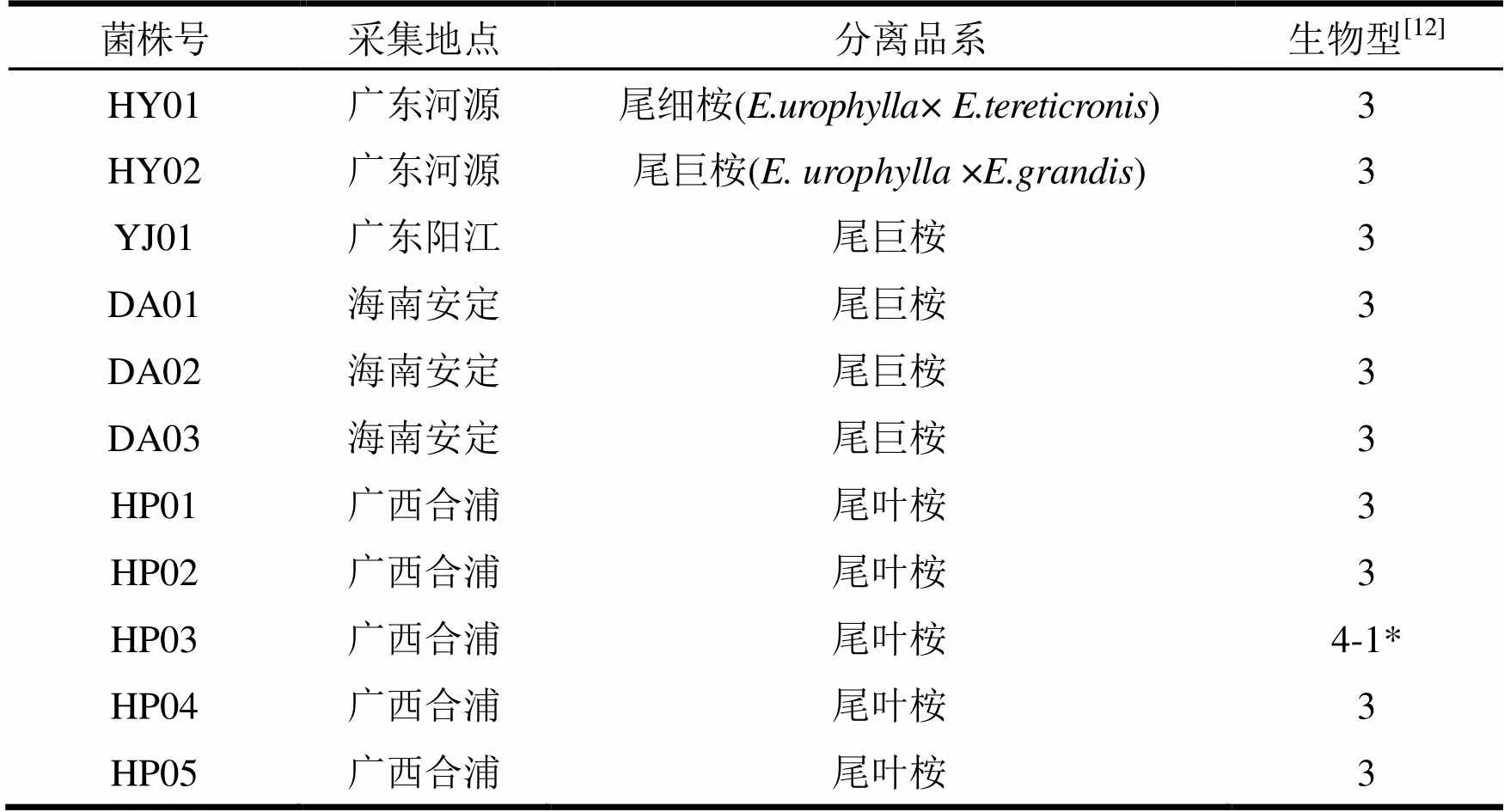
表1 供试菌株来源及其生物型
注:*表示青枯菌生物型采用苯酚红为指示剂的改良法测试[13],依照当前分类标准,无对应的生物型。这与曾宪铭等[14]在花生、姜和甘薯菌株中发现的情况相同,本研究中不确定生物型是首次在桉树无性系上发现。参照文献曾宪铭等标准,暂定为生化型Ⅳ的亚型(4-1)。
1.2 试验苗木
试验的尾巨桉无性系组培苗DH32-29由广东省湛江市南方国家级林木种苗示范基地苗圃提供。待幼苗长至株高约20 cm时,选择长势健壮组培苗木试验测试用。
1.3 试验方法
1.3.1 供试菌株的分子生物学鉴定
(1)病原菌基因组DNA的提取
纯化的菌株于CPG上28℃活化24 h,使TaKaRa宝生物有限公司的Lysis Buffer for Microorganism to Direct PCR提取病原菌基因组DNA。蘸取适量新鲜菌液置50 μL裂解液中,轻搅动几下后取出,80℃热变性15 min后低速离心,取上清液作为PCR反应模板。模板用的DNA以Nanodrop 2000微量紫外分光光度计检测纯度。
(2) PCR扩增
参照文献[15]以16sRNA为靶基因进行青枯菌特异性引物序列OLI1/Y2(OLI1:5’GGGGGTAGCTTGCTACCTGCC3’, Y2 :5’CCCACTGCTGCCTCCCGTAGGAGT3’)设计引物和合成[英潍捷基(上海)贸易有限公司]。PCR反应体系与琼脂糖凝胶电泳依据文献[16]实施。
(3)病原菌系统发育树的构建
PCR产物序列测定(华大基因公司)结果通过BLAST数据(http://blast.ncbi.nlm.nih.gov/Blast.cgi)同源性分析[17],以相似性较高(>98%)的序列作为同源序列,相邻属模式菌株16S rRNA序列作为外源序列(Outgroup)。MAFFT 7.0进行分子序列比对(软件具体设置为Scoring Maxtrix:1PAM/κ=2;Gap opening penalty=1.53;Offset value=0.0),序列比对完成后用BioEdit软件进行剪切编辑。以PAUP 4.0软件采用最大简约法(Maximum parsimony,MP)构建系统发育树,通过Bootstrap进行1000次可靠性检验,以Treeview软件对构建的发育树浏览。
1.3.2 病原菌的分离、纯化及菌悬液配制
病原菌分离培养基为TZC培养基(每升TZC培养基包含1 g水解乳蛋白、10 g蛋白胨、5 g葡萄糖和17 g琼脂,1%的2,3,5-氯化三苯四氮唑5 mL),调至适宜pH 6.5 ~ 7.0;于121 ˚C 高温灭菌20 min。
不同来源菌株致病性差异比较中的病原菌的分离、纯化及菌悬液配制如下:在酒精灯旁无菌环境下取常温保存的病原菌菌悬液一环,分别划线于TZC培养基,选择有强致病性的白色晕圈的粉红色菌落进行纯化培养48 h。培养好的菌落用无菌水洗脱菌体,10000×g离心10 min,去除上清液,无菌水梯度稀释后平板计数。并配制成108cfu·mL-1菌悬液保存备用。
1.3.3 桉树青枯病原菌致病性测试
2012年6月初,对分离纯化的11株菌株进行致病性测试。20 cm左右苗高的桉树组培苗先洗去基质,无菌水洗涤3次,消毒后的解剖刀距根尖1~ 2 cm处截断须根伤根,然后放入100 mL 108cfu·mL-1菌悬液中,每个处理30株组培苗,重复3次,无菌水对照。25 ~ 30°C温度及70% ~ 90%湿度、自然光照下培养10 d观察发病情况并统计试验期间的株数。在进行各试验指标统计之前,对病株作病原菌的分离鉴定,以排除非青枯死亡的影响,保证试验的准确性。
发病率计算参照文献[18]公式:发病率/% = (感染株数/总株数) × 100%。每天定时统计发病株数,计算日发病率,至试验结束时统计总发病株数并计算菌株的平均发病率。采用DataFit 7.1对每日发病率进行回归分析,根据回归模型确定试验期的发病率曲线情况和“发病高峰期(接种后到最大日发病率的天数)”。病原菌致病性(发病率经反正弦平方根处理值)采用方差分析和相关分析等方法统计各处理平均值的差异,并在SPSS 18.0数据处理系统中采用Duncan检验法对相关指标进行多重比较。
2 结果与分析
2.1 桉树病原菌16S rRNA基因序列鉴定
电泳结果如图1,以OLI1/Y2为特异性引物扩增16S rRNA基因序列,从采集的11株病原菌致病性测试发病桉树无性系组培苗中再次分离出病原菌后,均可在4 h内扩增出单一的特异性条带。测序结果与GenBank核酸数据库的Blast对比分析表明,供试病原菌均能与相似性高达99%。以PAUP 4.0软件采用最大简约法(Maximum parsimony,MP)分别对11株供试菌株与属中的5个16S rRNA测序的模式菌株进行系统发育树构建,结果如图2。11株病原菌和序列形成支持率为86%的分枝。系统发育分析也表明,11株病原菌16S rRNA 基因序列与具有高度同源性。
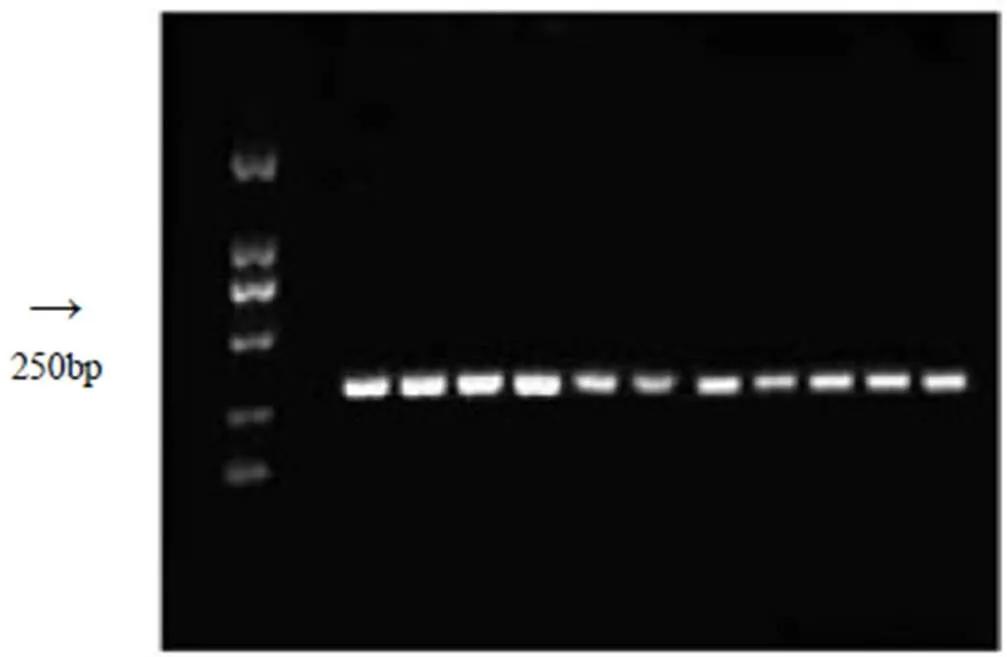
图1 11个菌株扩增后的电泳图谱
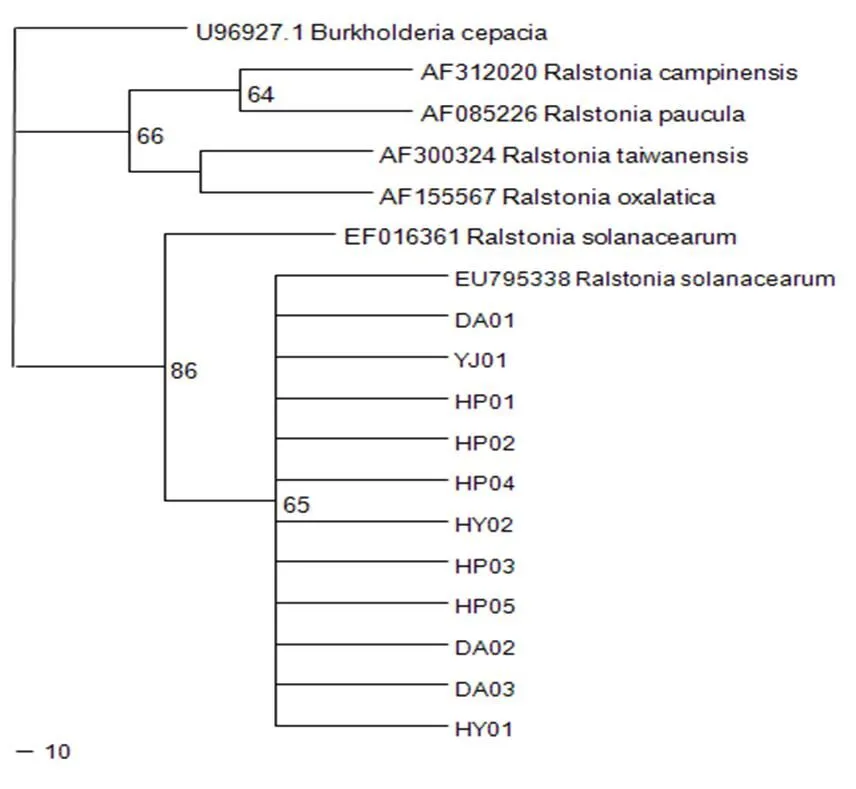
图2 11个菌株的分子系统发育树
2.2 桉树青枯病原菌致病性测试
桉树DH32-29苗通过伤根法接种病原菌后,均能在24 ~ 48 h内出现典型失水萎焉的青枯症状(图3),对照组不发病。通过对发病组培苗病原菌的形态及上述分子鉴定结果显示,进一步确定青枯症状由引起。

图3 桉树组培苗接种HY01处理后发病情况
注:图A-C:从左到右依次为无菌水对照组,接种菌株HY01处理和接种菌株YJ01处理。
进一步对不同菌株致病力进行了苗木测试,结果如图4和表2。桉树组培苗经不同菌株接种处理后,分别对桉树组培苗不同处理每天发病株数进行线性回归方程分析,结果发现不同菌株的发病率多集中在第4 天至第5 天表现出典型的感染症状(表3),其中YJ01处理最早达到发病高峰期(理论值为3.83 d),试验期内其处理平均发病率最低(40.00%),DA02处理苗木至发病高峰期的时间最长,超过5 d。总体而言,致处理桉树苗木平均发病率最大的菌株为DA03,平均发病率为86.67%,超过80%的菌株还有HY01、HP02、HP05。
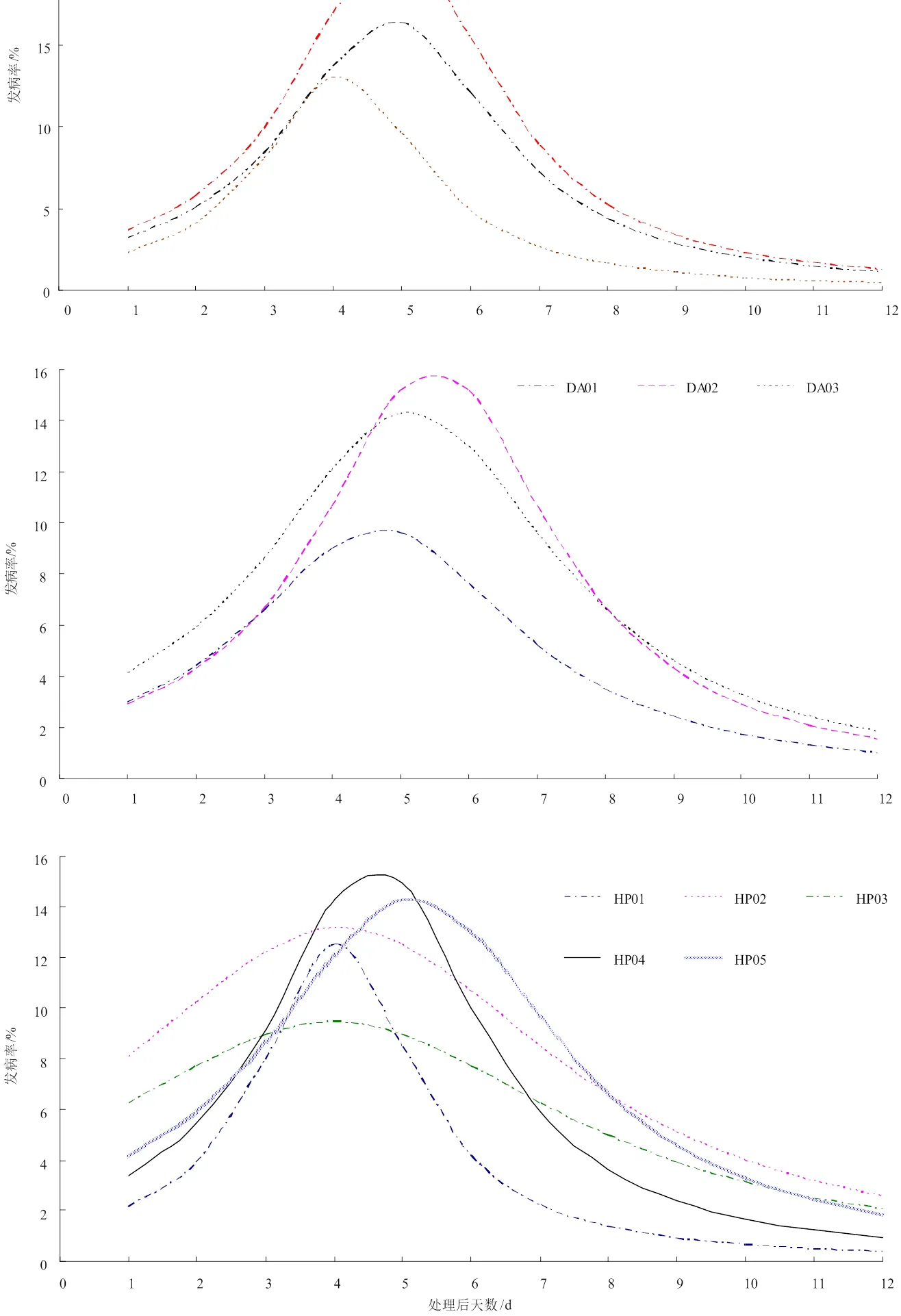
图4 桉树组培苗接种不同来源的菌株后日发病率情况率情况
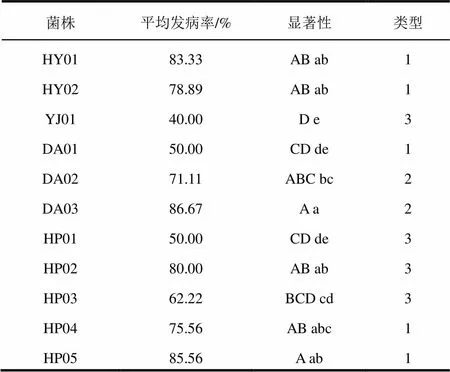
表2 不同菌株总平均发病率比较
注:显著性列中为平均值数值处理后比较结果,不同大写字母表示在0.01水平上差异极显著,小写字母表示在0.05水平上差异显著。下同。
以最后菌株的平均发病率值分别对测试菌株进行单因素方差分析,表明各病原菌在致病性(平均发病率值)上存在极显著的差异(<0.01),最低平均发病率的YJ01(40%)与最大平均发病率的DA03(86.67%),HP03(生物型4-1)菌株与DA03、HP05菌株在致病性表现出极显著差异(< 0.01),同时HP03与HY01、HY02、YJ01、DA03、HP02、HP05均表现出0.05水平上的显著差异。采用K-均值聚类分析法(K=3),以发病高峰期、高峰期发病率和平均发病率为变量,对不同地理来源菌株间致病性差异比较,结果表明HY01、HY02、DA01、HP04和HP05聚为第1类,DA02和DA03聚为第2类,其余为第3类(表3)。对4个不同来源(采样点)的菌株平均发病率进行方差分析,结果表明,来源广东阳江菌株与其他3个来源地菌株在致病性上差异显著(图5)。

图5 不同来源菌株致病性比较分析
3 讨论与结论
由劳尔氏菌引起的青枯病具有遗传多样性,其寄主、地理分布、致病性以及生物型均有差异[19]。本研究结果表明,分离获得的病菌菌株进行致病性的测试,接种桉树组培苗试验后,观察到的发病症状与林间自然发病一致,尽管不同种源的菌株之间在致病力中存在着显著差异,但经生化型测定[12]以及16S rDNA序列分析鉴定,桉树青枯病均由病原菌引起,也说明不同种源以及同一种源内的存在着地理分布、致病性以及生物型等方面的差异。已有的研究表明我国的桉树青枯病菌株的主要为生理型3,与其宿主的互作机制中,青枯病菌属于分泌蛋白型细菌( proteobacteria)的III型(Type III Secretion System,T3SS)[20],而青枯病病菌致病力大小与保守T3SS序列中作用的效应因子类型有关[21]。
本研究结果发现菌株致病力与地理来源无明显的关联性,即使具有高度同源的16S rRNA基因,来源于同一区域菌株在致病力存在着差异,不同生物型致病性亦无显著性差异,这也表明不同地理区域的青枯菌在与其寄主长期协同进化的过程中,会演化出明显的生理分化或菌系多样性,这些可能与其T3SS效应因子的Rip (protein injected into plant cells)基因多样性密切相关[21]。本研究仅对1个桉树无性系进行致病性测试,不同地理来源菌株对其他桉树品系的致病力是否一致,有待于进一步研究。综上所述,桉树青枯菌种群组成较复杂。因此在生产实践中对桉树青枯病生物防治时,应充分了解各桉树栽培区病菌的致病力等方面的差异以及其作用机制,以此有针对性地进一步的生物防治、抗性桉树无性系选育等试验研究。
[1] Hayward A C,Biology and epidemiology of bacterial wilt caused by[J].Annual Review of Phytopathology,1991,29(1):65‒87.
[2] 何学友.我国林木青枯病研究概况[J].森林病虫通讯,1997(1):43‒46.
[3] 曹季丹.巴西柳桉、巨桉青枯病调查初报[J].广西林业科学,1982(4):30‒31.
[4] Xu J,Pan Z C, Prior P,et al.Genetic diversity ofstrains from China[J].Plant Pathology, 2009,125 (4):1‒13.
[5] 林绪平,张民兴,许少嫦.广东桉树青枯病的发生特点及防治特性[J].广东林业科技,1995,11(1):52‒53.
[6] Bernard D, Xu D P. Bacterial wilt and boron deficiency stress: a new disorder in eucalypt plantations in south China[J]. Chinese Forestry Science and Technology 2006,5(1): 45–50.
[7] Xu J, Pan Z C, Prior P, et al. Genetic diversity ofstrains from China[J]. European Journal of Plant Pathology,2009,125(4):641‒653.
[8] Genin S, Boucher C.: secrets of a major pathogen unveiled by analysis of its genome[J]. Molecular Plant Pathology,2002,3(3): 111‒118.
[9] 伍慧雄,王胜坤,孙思.桉树青枯病的发生与防治[J].广东林业科技,2006,22(3) :53‒56.
[10] 吴清平,梁子超.桉树青枯病病原鉴定和致病力测定[J].华南农业大学学报,1988,9(3): 59‒67.
[11] 王胜坤.桉树青枯菌菌株致病力分化及吸附识别及PCR快速检测研究[D].北京:中国林业科学研究院, 2007.
[12] Wang Y, Arnold R, Li G Q, et al. Identification and rapid detection of bacterial wilt in plantationin China[J]. Australian Forestry,2014,77(2):133‒139.
[13] Huang Q,Yan X R,Wang J F.Improved biovar test for[J].Microbiological ethods,2012,
88(2):271‒274.
[14] 曾宪铭,董春.广东农作物青枯病的生物型[J].华南农业大学学报,1995,16(1):50‒53.
[15] Seal S E, Jackson L A., Young J P,et al. Differentiation of,,and the blood disease bacterium by partial 16S rRNA sequencing: construction of oligonucleotide primers for sensitive detection by polymerase chain reaction[J].Journal of General Microbiology,1993,139(7): 1587–1594.
[16] 王艳,吴志华,李国清,等.桉树组培苗潜伏期和轻基质中青枯菌的PCR快速检测[J].中南林业科技大学学报,2013,33(9):42‒45.
[17] Salanoubat M, Genin S, Artiguenave F, et al. Genome sequence of the plant pathogen[J].Nature,2002,415: 497–502.
[18] 方中达.植病研究法(3版)[M].北京:中国农业出版社,1998.
[19] Monteiro F, Solé M, Van D I, et alA chromosomal insertion toolbox for promoter probing, mutant complementation, and pathogenicity studies in[J].Molecular Plant‒Microbe Interactions, 2012, 25(4): 557‒568.
[20] 吴志华,谢耀坚,罗联峰,等.我国桉树青枯病研究进展[J].林业科学研究,2007,20(4):569‒575.
[21] Wang K,Remigi P,Anisimova M, et alFunctional Assignment to Positively Selected Sites in the Core Type III Effector RipG7 from[J]. Molecular Plant Pathology,2016,17(4):553‒564.
Molecular Identification and Pathogenicity Bacterial Wilt Isolated from DifferentClones
WANG Yan1,2, XIE Yao-jian1, LI Guo-qing1, WU Zhi-hua1
11isolates obtained from differentclones were used to identify phylogeny and compare differences in pathogenicity. Tissue culture seedlings of the hybrid eucalypt clone DH32-29 were used in pathogenicity tests, which involved infection by dipping of wounded roots. The results of phylogeny showed that the 16S rRNA sequences of all 11 isolates were highly homologous tothe type strain ofwith86% bootstrap support, and all belonged to the same phylotype. There were significant differences in wilt incidence among 11 isolates (<0.01 to<0.05). Based on three features of the isolates – fastigium, pathogenicity of fastigium and mean morbidity – the isolates were segregated into 3 groups by clustering on k-means: HY01, HY02, DA01, HP04 and HP05 formed one group; DA02 and DA03 formed a second group; and, YJ01, HP01, HP02 and HP03 formed a third group. One-way analysis of variance (ANOVA) was used to compare wilt pathogenicity of the 11 isolates, which had been collected from four different locations; the results showed that an isolate collected from Yangjiang, Guangdong province, differed significantly (<0.01) to all others in this respect. In view of all factors, there was no significant difference (<0.05) in wilt incidence between 2 of the biovars (biovar 3 and biovar 4-1), which had been obtained from the same plantation. The results from this study provide technical support to both efforts to prevent and control the occurrence ofbacterial wilt and also to the breeding of resistant eucalypt varieties for the eucalypt industry in southern China.
bacterial wilt; molecular identification; pathogenicity; biovar
S763.13
A
“十二五”农村领域国家科技计划专题“南方短周期工业用材林病虫害控制技术”(2012BAD19B0802)
王艳(1986—),硕士,助理工程师,主要从事森林可持续发展研究.
吴志华(1974—),硕士,副研究员. E-mail:wzhua2889@163.com
猜你喜欢
儿童故事画报·发现号趣味百科(2019年9期)2019-02-02 04:12:19
环球时报(2019-01-03)2019-01-03 09:06:44
现代园艺(2018年3期)2018-02-10 05:18:20
西南农业学报(2016年6期)2016-04-16 05:12:49
广西林业科学(2016年2期)2016-03-20 05:53:22
广西林业科学(2016年4期)2016-03-16 05:44:51
现代农业(2015年1期)2015-02-28 18:39:51
新疆农垦科技(2014年6期)2014-02-28 19:20:13
当代畜禽养殖业(2014年3期)2014-02-27 07:58:51
植物营养与肥料学报(2012年5期)2012-10-26 03:28:36
