
一种基于连锁不平衡的tagSNPs选择算法
2016-02-27 07:18:39王钧峰王新赠
泰山学院学报 2016年3期
王钧峰,王新赠
(山东科技大学数学与系统科学学院,山东青岛266590)
一种基于连锁不平衡的tagSNPs选择算法
王钧峰,王新赠
(山东科技大学数学与系统科学学院,山东青岛266590)
进行全基因组关联研究(genome-wide association studies,简记为GWAS)时,我们需要获得一个足够密集的单核苷酸多态性(single-nucleotide polymorphism,简记为SNP)标记集来解释常见疾病遗传风险的一部分.候选基因中SNP的数量是有限的,但是直接分析所有现存的SNPs是无效的,因为在这些位点上的基因型有很强的关联性,会导致大量的冗余信息,并且会造成基因分型成本的增加,消耗大量的时间.所以我们在进行关联检验时,没有必要对所有的SNPs进行基因分型,只需要选择出具有代表性,并且数量很少的SNPs进行分型,并对这些SNPs进行关联检验.这里选择出的SNPs称为标签SNPs(记为tagSNPs),它为SNPs的一个小的子集,在每个单体型区域中足以捕获单体型的信息.选择tagSNPs的方法有很多,本文我们提出了一种新的tagSNPs选择方法,通过使用基于连锁不平衡(linkage disequilibrium,简记为LD)的两两r2准则对单体型分组,分成不相交的组,并在每个组中选择标签SNPs.与基于原始SNPs集的检验方法比较,我们的方法产生了更少的tagSNPs,在最大化所选标记提供信息含量的同时,降低了基因分型成本,提高了效率.
全基因关联研究;SNP;标签SNP;连锁不平衡
1 引言
在人类基因组中,由于SNP高的丰富性,低突变率,易于高通量基因分型[1],所以SNP在疾病关联研究中起到了很重要的作用.TagSNPs的选择已经成为一个非常活跃的研究课题.如果能在SNP数据集中选择出tagSNP集,就可以减少用于关联检验所使用的SNP的数量,这样也就降低了基因分型的成本和计算的时间复杂度.目前已经被提出的tagSNP选择方法主要有两种,分别是基于单体块结构的识别方法[2-5]和基于LD的识别方法[6-8].很多算法也已经被提出来检测单体型块和标记的选择.Patil[9]等定义一个单体型块,单体型块是一个区域,在其中所有观察的a%的单体型是常见单体型,常见单体型是单体型频率大于某一阈值的单体型,通过这个方法选择所有可能的单体型块,然后通过一个贪婪优化算法选择出连续没有重叠的单体型块和tagSNPs.Johnson[2]等是基于连锁不平衡计算两两SNPs间的连锁不平衡程度,如果连锁不平衡程度大于某个阈值,那么其中一个就可以作为tagSNP.Zhang[3]等使用一个动态编程方法来进行单体型块的划分和tagSNP的选择.Cardon[10]的策略是选择一个具有代表性的SNPs集,不考虑剩余SNPs,目的是保留大多数原始集信息的同时,所选择的SNPs没有信息的重叠.Carlson[8]利用贪婪算法识别tagSNP,从所有超过某阈值的SNPs开始,与最大数量SNPs之间的连锁不平衡程度都大于某个阈值的SNP作为tagSNP.Zhang[5]等人,介绍基于LD的方法,这些方法搜索一个小的SNP集,并与其他不被选择的SNPs有强的连锁不平衡.Stram[11]等给出了一种统计方法,在其中多个tagSNPs可以被用来代表每个没有被标记的SNP.
在应用我们提出的方法选择tagSNPs之后,我们要验证所选择的tagSNPs是否可以验证与疾病的关联性,所以我们要进行假设检验.研究者已经提出了很多检验方法.CAST[12]是对于每个个体把在一个区域内(例如,一个基因的外显子)所有罕见变体信息重叠成一个二分变量,通过判断个体是否有任何罕见变体在这区域内,然后运用一个单变量检验[13].CMC[14]扩展了CAST方法,通过在一个等位基因频率的基础上,把罕见变体重叠在分组的区域内,重叠所分的组就如CAST方法一样,并对所分的组用一个多元检验.加权和检验(WST)[15]考虑一组病例对照,把一个SNP集重叠成一个罕见等位基因数量的单一的加权平均.Wu[16]提出了一种基于逻辑核机器的逻辑核检验方法(sequence kernel association test,简记为SKAT),SKAT假定标记的回归系数的一个分布,其方差取决于灵活的权重.SKAT执行一个基于得分的方差分量检验,它的计算只需要拟合空模型通过单独在协变量上回归表型和解析计算P值.SKAT能够直接获得一个P值而不需要排列求P值.SKAT的一个重要特性是它允许结合灵活的加权函数来提高分析功效.所以在进行假设检验时,我们使用高效灵活的SKAT.
2 方法
2.1 tagSNP的选择
考虑包含P个双等位基因SNP标记a1,a2,…,αp的一个集.进一步假设所有这些标记次要等位基因频率(MAF)超过一个特定的阈值(在这使用0.05).首先,计算两两LD测量r2[17].如果两个标记ai和aj间的r2大于一个特定的阈值r0,那么就说它们两个有强的LD,表示为r2(ai,aj)≥r0(在这个研究中r0=0.8),两个都可以被考虑作为对方的tagSNPs,在其中ai可以用来作为aj的一个替代,反之亦然.
我们的目的是找到一个tagSNP集,对于基因分型,我们开发了一种算法来识别tagSNPs子集,从超过一个给定MAF阈值的所有SNPs中选择.从超过MAF阈值的所有SNPs开始,对我们所选择的基因区域进行分组,分成几个SNP子集,SNP集中任意两个SNPs同属于一个组当且仅当这两个SNPs之间的r2大于等于给定的阈值,也就是说同一个组中的SNPs至少与同组中的一个SNP连锁不平衡,分组的过程是迭代的,每一次循环分析所有未被分组的SNPs,直到所有的SNPs被分组,这样,就分成了几个组,但是会出现这样一种特殊情况,有的SNPs和任何SNPs都不连锁不平衡,那么我们就把这样的单个SNP单独作为一个组.
这样,组中一个SNP被指定为“tagSNPs”,每个组只有一个tagSNP将会需要被基因分型.下面,我们就在每个组中选择一个tagSNP,再把每个组中选择的tagSNP组成总的tagSNP集,然后进行假设检验.选择tagSNP的方法具体如下,在一个已经分好的组中进行选择,我们首先计算出组中最大r2值的两个SNPs,然后再从这两个SNPs中选择其中一个作为tagSNP,分别计算这两个SNPs与其他除去二者本身的组内剩余SNPs的r2值的和,哪个值大,我们就选择哪一个来作为tagSNP,如果相等那么我们就任选其中一个来作为tagSNP,我们选择的研究对象没有出现这种情况.
2.2 r2的计算
给出m个个体,m/2个病例和m/2个对照,所有P个位点上的单体型Zij∈{0,1},i=1,2,…,2m,j =1,2,…,P.计算生物学中描述SNP间相关关系的连锁不平衡系数[18]r2:
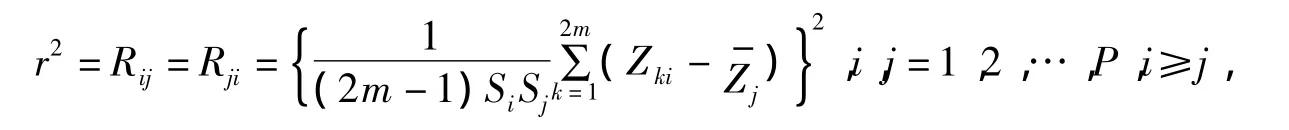
2.3 SKAT
现有n个独立个体,对于给定的含s个SNPs的SNP集,设Zi1,Zi2,…,Zis,是第i个个体在这s个SNPs上的基因型值,i=1,2,…,n.显然,Zij∈{0,1,2}.第i个个体的定性性状用yi表示,若个体i患病,则yi=1,否则yi=0.
下式(1)给出了个体定性性状和基因型值间的半参数模型
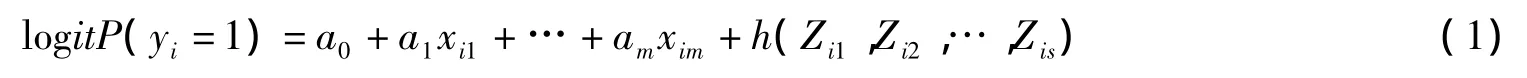


本文使用SKAT检验方法,分别对基于原始SNP集和基于标签SNP集进行检验.基于原始SNP集的检验用SKAT表示,基于标签SNP集的检验用SKAT-tag表示.然后比较二者的P值和功效.
3 仿真研究
3.1 仿真数据
为了计算对两种SNP集检验的P值和功效,我们进行了大量的仿真实验,本文的仿真数据均由HAPGEN2[22]产生,并且事先假定所有原因SNPs都会增加致病风险.第13号染色体携带许多与乳腺癌有关的基因,我们选择其中的MTRF1基因,它包含62个HapMap[23]SNPs.这62个SNPs中的10个SNPs已经由Illumina HumanHap 500 array给出了确定的基因型.我们使用HapMap上的CEU样本,用HAPGEN2基于CEU样本的连锁不平衡结构产生仿真数据.
我们使用HAPGEN2在不同的参数下产生MTRF1基因上62个SNPs的1000组仿真数据,每组包含500个病例和500个对照.我们从这100组中随机选择1组并从该组中随机选取50个病例和50个对照的200条单体型数据上,在这200条单体型数据上运用我们的方法选取tagSNPs,之后所有的仿真实验和假设检验都以现在选取的SNPs作为tagSNPs.
3.2 P值计算
我们使用来自HapMap计划中174个CEU种族个体的真实单体型数据,分别对tagSNPs集和原始SNPs集进行关联检验,我们在显著水平a=0.05下使用SKAT检验方法,求其P值,见表1.

表1 P值
使用原始SNPs集,求得的P值为0.037,远小于0.05,所以使用原始集在统计学上显著关联.使用我们方法选择的tagSNPs集,所求的P值为0.048,也小于0.05,所以我们的方法选择的SNPs集在统计学上微弱显著关联,因为我们的方法选择了少数的SNPs,所以丢失很多信息,我们的方法微弱显著性关联,说明我们选择的tagSNPs具有代表性,也说明我们方法选择的tagSNPs可以用来进行疾病关联检验.虽然我们的方法不如使用原始集关联显著,但是我们方法计算速度明显高于基于原始集的方法.
3.3 第I类错误率估计
利用HAPGEN2产生空模型下的1000组仿真数据来估计第I类错误率,每组方针数据包含500个病例和500个对照.如表2得到的第I类错误率,显著水平为a=0.05.SKAT方法和SKAT-tag方法的第I类错误率分别为0.049和0.042.说明SKAT检验方法都能很好地控制第I类错误率.

表2 第I类错误率
3.4 功效估计
我们将基因MTRF1上由Illumina HumanHap 500 array给出的10个已确定基因型的SNPs每个轮流作为致病SNP,并有HAPGEN2进行仿真,10个已确定基因型的SNPs每个轮流一次就仿真1000组,总共为10000组,我们假定杂合子致病风险为1.25,纯合子致病风险为1.5.表3给出了MTRF1基因上已确定基因型的SNPs.

表3 MTRF1基因上已确定的10个SNPs
我们将表3给出的10个SNPs中每个SNPs轮流作为致病SNP,每一个SNPs作为致病SNPs,分别使用SKAT和SKAT-tag求一次功效,显著水平a=0.05,然后比较两种方法,10个SNPs各轮流作为致病SNPs的功效,如图1所示.
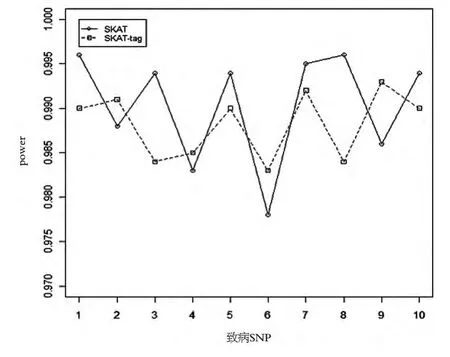
图1 10个致病SNP分别在使用SKAT和SKAT-tag方法时的功效
4 结果分析和讨论
在本文中我们提出了一种tagSNPs的选择方法,降低了基因分型的成本和计算的时间复杂度.与原始SNPs集相比,我们不需要对所有的SNPs进行基因型的测定,也不需要对所有SNPs进行关联检验,而只需要对我们选择的tagSNPs进行基因分型和关联检验.通过我们的方法对MTRF1基因上62个SNPs进行tagSNPs的选择,最终我们选择了rs666930,rs586650,rs550174,rs483180,rs616111,rs6668589,rs523395,rs2246410,rs512854这11个tagSNPs,数量大约为原始SNPs集的1/6,基因分型的成本也就降低了5/6,P值的计算时间也会减少.因为我们首先在一小部分数据中选择tagSNPs,所以总的来说,使用tagSNPs进行关联检验的时间复杂度比使用原始SNPs集进行关联检验的时间复杂度要小.
通过使用来自HapMap的174个CEU种族个体数据,我们对两种SNPs集使用SKAT检验方法求得P值,如表1所示,基于tagSNPs集检验的P值小于基于原始SNPs集检验的P值.但是二者的P值都小于显著性水平,也就两种SNPs集对疾病都显著性关联,所以我们的方法求得的tagSNPs可以用来进行疾病关联检验.从表2可以看出,SKAT对第I类错误率都是可控的.图1表明基于原始SNPs集检验的功效与基于tagSNPs集检验的功效在大部分情况下相差不大.但在某些情况下,基于tagSNPs集检验的功效小于基于原始SNPs集检验的功效.还有使用tagSNPs集得到P值小于使用原始SNPs集得到的P值,很大程度上是因为关联检验时没有包含所有SNPs,所以造成了信息的大量丢失.某些情况我们方法功效大于原始SNPs集方法,可能因为SNPs数量的减少,造成了自由度的降低.总的来说,我们的方法计算复杂度低,基因分型成本低,是可以用来进行疾病关联检验.
[1]Collins F.S,et al.Variations on a theme:cataloging human DNA sequence variation[J].Science,1997(278):1580-1581.
[2]G.C.Johnson,L.Esposito,B.J.Barratt,et al.Haplotype tagging for the identification of common disease genes[J].Nature Genetics,2001(2):233-237.
[3]K.Zhang,M.Deng,T.Chen,M.S.Waterman,F.Sun.A dynamic programming algorithm for haplotype block partitioning[J].Proceedings of the National Academy of Sciences of the United States of America,2002(11):7335-7339.
[4]E.C.Anderson,J.Novembre.Finding haplotype block boundaries by using the minimum-description-length principle[J].American Journal of Human Genetics,2003(2):336-354.
[5]K.Zhang,P.Calabrese,M.Nordborg,F.Sun.Haplotype block structure and its applications to association studies:power and study designs[J].American Journal of Human Genetics,2002(6):1386-1394.
[6]Weale M.E.,et al.Selection and evaluation of tagging SNPs in the neuronal-sodiumchannel gene SCN1A:implications for linkagedisequilibrium gene mapping[J].Am.J.Hum.Genet,2003(73):551-565.
[7]Ke X.,Cardon L.R.Efficient selective screening of haplotype tag SNPs[J].Bioinformatics,2003(19):287-288.
[8]Carlson C.S.,et al.Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium[J].Am.J.Hum.Genet,2004(74):106-120.
[9]N.Patil,A.J.Berno,D.A.Hinds,et al.Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21[J].Science,2001(5547):1719-1723.
[10]Cardon L.R.,Abecasis,G.R.Using haplotype blocks to map human complex trait loci[J].Trends Genet.,2003(19):135-140.
[11]Stram D.O.,et al.Choosing haplotype-tagging SNPs based on unphased genotype data using preliminary sample of unrelated subjects with an example from the multiethic cohort study[J].Hum.Hered.,2003(55):27-36.
[12]Morgenthaler S.,Thilly W.G.A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases:a cohort allelic sums test(CAST)[J].Mutat.Res,2007(615):28-56.
[13]Morris A.P.,Zeggini E.An evaluation of statistical approaches to rare variant analysis in genetic association studies[J].Genet.Epidemiol,2010(34):188-193.
[14]Li B.,Leal S.M.Methods for detecting associations with rare variants for common diseases:application to analysis of sequence data[J].Am.J.Hum.Genet,2008(83):311-321.
[15]Madsen B.E.,Browning S.R.A groupwise association test for rare mutations using a weighted sum statistic[J].PLoS Genet,2009(5): e1000384.
[16]Wu M C,Kraft P,Epstein M P,et al.Powerful SNP-set analysis for case-control genome-wide association studies[J].The Ameri-can Journal of Human Genetics,2010,86(6):929-942.
[17]Devlin B,Risch N.A comparison of linkage disequilibrium measures for fine-scale mapping[J].Genomics,1995(29):311-322.
[18]Hill W G,Robertson A.Linkage disequilibrium in finite populations[J].Theoretical and Applied Genetics,1968,38(6):226-231.
[19]Liu D,Ghosh D,Lin X.Estimation and testing for the effect of a genetic pathway on a disease outcome using logistic kernel machine regression via logistic mixed models[J].BMC bioinformatics,2008,9(1):1-11.
[20]Zhang D,Lin X.Hypothesis testing in semiparametric additive mixed models[J].Biostatistics,2003,4(1):57-74.
[21]Lin X,Cai T,Wu M C,et al.Kernel machine SNP‐set analysis for censored survival outcomes in genome‐wide association studies[J].Genetic epidemiology,2011,35(7):620-631.
[22]Su Z,Marchini J,Donnelly P.HAPGEN2:simulation of multiple disease SNPs[J].Bioinformatics,2011,27(16):2304-2305.
[23]The International HapMap Consortium.The International HapMap Project[J].Nature,2003(426):789-796.
The Method of Selecting tagSNPs Based on Linkage Disequilibrium
WANG Jun-feng,WANG Xin-zeng
(School of Mathematics and Systems Science,Shandong University of Science and Technology,Qingdao,266590,China)
In genome-wide association studies,we need to have a sufficiently dense single nucleotide polymorphisms set to explain part of the genetic risk for common diseases.Within candidate genes,the number of common polymorphisms is finite,but direct assay of all existing common polymorphism is inefficient,because genotypes at many of these sites are strongly correlated,can lead to a large amount of redundant information,and will result in an increase in the cost of genotyping,consume large amounts of time.So when we test the association of markers with disease,typing all available SNP markers is inefficient and not necessary.We only need to select a representative,small number of SNPs for genotyping,and test the association between these SNPs and disease.The SNPs selected here called tagSNPs,it is a small subset of the SNPs,and enough to capture the haplotype information in every haplotype region.The selection of tagSNPs has become a very active research topic and many strategies have been proposed.In this paper,we put forward a new kind of tagSNPs selection method,by using measure based on pairwise LD to group the haplotype,divided into disjoint groups,and selected the tagSNPs in each group.Compared with testing method based on original SNPs sets,our method has produced less tagSNPs,while simultaneously maximizing the information content by selected markers,reducing the cost of genotyping,and improving the efficiency.
genome-wide association studies;SNP;tagSNPs;linkage disequilibrium
Q811.4
A
1672-2590(2016)03-0049-06
2016-04-03
国家自然科学基金资助项目(61572522)
王钧峰(1990-),男,河北沧州人,山东科技大学数学与系统科学学院硕士研究生.
猜你喜欢
基层中医药(2020年5期)2020-09-11 06:32:00
现代装饰(2020年7期)2020-07-27 01:27:44
基层中医药(2018年5期)2018-08-31 02:35:42
NBA特刊(2018年7期)2018-06-08 05:48:32
现代装饰(2018年4期)2018-05-22 02:57:23
中国军转民(2017年7期)2017-12-19 13:30:00
大连工业大学学报(2015年4期)2015-12-11 04:06:50
制造技术与机床(2015年10期)2015-04-09 07:06:14
汽车维护与修理(2015年6期)2015-02-28 12:16:55
中国卫生(2014年10期)2014-11-12 13:10:24
